阅读:0
听报道
编者按
病毒突变不是什么新鲜事,尤其是RNA病毒,突变更是常见,这也是为什么流感病毒疫苗相对于乙肝病毒疫苗来说难以研发的因素之一,因为前者是RNA病毒,后者是DNA病毒。对于每一个接触过分子生物学实验、有着基本科学素养的人来说,“突变”更像是一种中性词,有好的一面,也有坏的一面。然而,对于大多数非专业领域的人员来说,则谈“突变”色变。当然,一方面需要科研人员带来更权威、更专业的解释(需要提一句的是,我们都知道科研更多的是一种“提出假说-验证假说-否定假说-再提出新假说”的过程,因此我们也应该给科研人员更多的宽容,因为本来科研就是不断完善和发展的),另一方面,在这个人人都是媒体的年代,如何能坚持底线,不盲目解读科学结论、不传播不责任的内容,也是每一个在传播链节点上的个体要思考的。
撰文 | 唐小糖 责编 | 兮
SARS-CoV-2自暴发以来,“突变”始终是人们所关心的话题,截至目前已有数十篇研究聚焦此话题。以最近的几篇研究为例,2020年2月17日,广东省疾控中心在预印本medRxiv的一篇研究表明广东的SARS-CoV-2和武汉早期的SARS-CoV-2 基因组之间几乎没有发生突变[1];2月19日,中科院西双版纳热带植物园在预印本ChinaXiv上传的研究通过分析93个样本的基因组数据发现仅有120个核苷酸位点发生突变,这些突变均匀分散在10个编码区[2];2月25日,南方医科大学分析比较38株病毒基因序列后发现,以最早测定的序列为参考,共计有117个位点突变,变异位点没有聚集性,相对均匀地分布整个基因组[3]。
3月2日,加州大学洛杉矶分校程根宏、中国CDC谭文杰、苏州系统医学研究所蒋太交、中国CDC武桂珍等合作在预印版平台bioRxiv发表了文章Mutations, Recombination and Insertion in the Evolution of 2019-nCoV,分析了包含11株新收集于中国病人的病毒序列在内的120条新冠状病毒基因组序列,追踪了多个继承性单核苷酸多态性位点(SNPs),确定了新冠病毒与其他冠状病毒之间的简化关系。该研究也发现基因组中仅有几个突变,整体突变程度较低。研究人员还发现了两个具有不同SNPs标记的遗传亚群的共流行:尽管新冠状病毒、人SARS病毒和蝙蝠SARS病毒在基因组结构上具有高度同源性,但它们通过受体结合区的潜在重组,进化成具有不同受体进入特性的两个不同群体。研究结果暗示,新型冠状病毒可能通过受体结合区重组和furin或TMPRSS2裂解位点插入增加其感染性。这一发现表明当前病毒分为两个类型,且两个类型在病毒暴发的早期共存,可用于追踪新冠状病毒在不同地区和国家的传播途径[4]。(详见《独家全文编译 | 新型冠状病毒在进化过程中的突变、重组和插入》)
以上各项研究表明在约30000个碱基的病毒基因组中,目前整体突变程度较低,未发生重组现象。
2月18日,耶鲁大学流行病学专家Nathan D. Grubaugh等专门在Nature Microbiology上发表评论文章,指出病毒突变符合正常流行病学规律,不应该引起恐慌[5]。
2020年3月3日,北京大学生命科学学院陆剑课题组和中国科学院上海巴斯德研究所崔杰课题组合作,在National Science Review在线发表题为“On the origin and continuing evolution of SARS-CoV-2”的文章,对SARS-CoV-2的演化动态进行新的解读。不同的突变类型具有不同的演化压力,非同义突变率与同义突变率的比率,ω = dN/dS,用以描述在蛋白质水平上的选择压力。该研究首次考虑到非同义突变受到负选择(淘汰)的压力大,并通过对比同义(中性)突变来研究SARS-CoV-2的演化进程。并且根据基因组不同的突变位点,将SARS-CoV-2的分为L和S两个类型,同时探讨了当前人为干预措施对两种类型演化动态可能产生的影响。

研究人员分析了来自公共数据库的103个SARS-CoV-2基因组,结果表明这些病毒株一共存在149个突变位点,且多数为近期发生。进一步分析发现位于参考基因组 (NC_045512) 第8782位的T-C突变(同义突变)和28144位的C-T非同义突变的两个突变位点高度连锁。在所分析的103个SARS-CoV-2基因组中,有72个(约70%)基因组在28144位ORF8基因中对应的氨基酸为亮氨酸(L),定义为“L”型;29个 (约28%)在28144位对应的氨基酸为丝氨酸(S),定义为“S”型,另外2个基因组中,1例可能是L和S型的混合体,而另1例突变则不属于这两个类型。通过与其他冠状病毒进行比较,发现S型与蝙蝠冠状病毒在进化树上更接近,更古老可能为祖先类型(在当前病毒溯源不是十分清楚的情况下不能绝对化),L型则可能由S型演化而来。
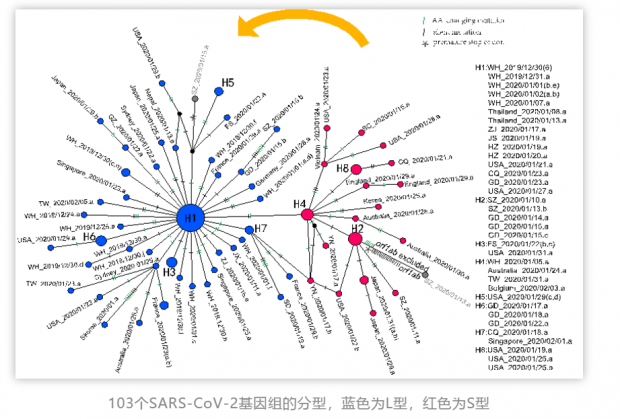
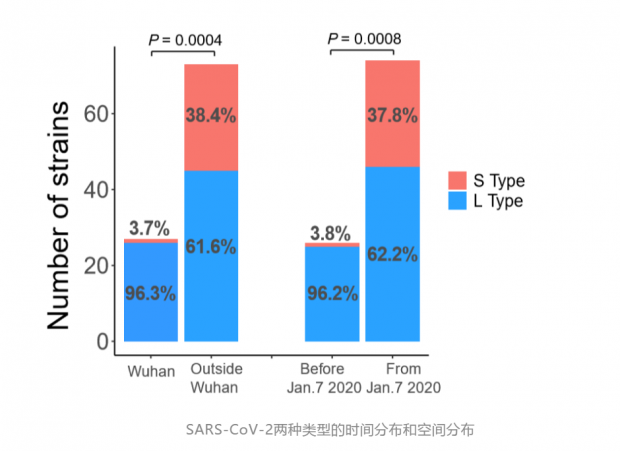
L和S两种类型在空间和时间上分布各有什么特点?在所分析的病毒中,来自武汉的病人中,有26 例(96.3%)感染的是L型,1例(3.7%)感染的是S型;而武汉外的患者有45 例(61.6%)感染的是L型,28例(38.4%)感染的是S型。这表明武汉患者主要感染的是L型;时间上来看,在1月7日前,采样全部来自武汉,S型比例不到4%,但1月7日之后,绝大部分样品来自武汉之外,随着病毒在人群中的持续传播,S型的比例逐渐上升至38%。
在早期的传播过程中,作为祖先型的S型几乎完全被L型取代,但随着时间推移,S型又逐渐上升,研究人员推测L型可能传播能力更强,复制速度更快,潜伏期更短,因而早期会在人群中快速扩张。但随着医疗和隔离措施的加强,L型的负选择压力变大,所以S型频率逐渐上升。
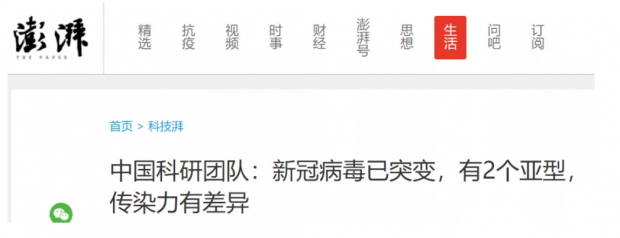
读者们尤其要注意的的一点是,不管是L型还是S型(且不说仅仅是基于有限数据的推论),其中之一都不是在在人群中传染过程中显著变异,而更有可能是暴发时候就存在。这一点,澎湃新闻存在明显的误读(上图),让大众误以为病毒经过这一两个月的传播在人群传播过程中发生明显变异导致复制速度更快和潜伏期更强。新冠病毒作为RNA病毒突变有什么大惊小怪?传染力有差异也仅仅是推测。大众媒体下结论一定要慎之又慎。该话题微博占据热搜第一很长时间,阅读量超过4亿,不少网友留言表示惊恐。这样的新闻实在是要不得。
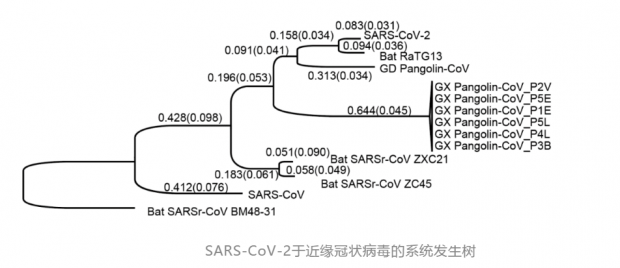
此外,研究人员还分析SARS-CoV-2与蝙蝠冠状病毒RaTG13的基因组差异,发现虽然SARS-CoV-2与蝙蝠冠状病毒RaTG13总体只有约4%的差异,但同义突变的差异却达到17%。这一结果说明两者的遗传距离比之前的认识实际上要远得多,相当于人与黑猩猩差别的14倍,人与恒河猴差别的2倍。SARS-CoV-2 和来自广东的穿山甲冠状病毒之间的基因组的dS值为 47.5%,这与人类和小鼠的差异(50%)相当,甚至SARS-CoV-2和广西穿山甲冠状病毒的差异更大,为72.2%。
研究人员通过对比广东的马来穿山甲冠状病毒发现,S蛋白的RBD区域具有较高的突变率(F486、Q493、 S494、 N501和Y505五个关键位点dS = 0.411、dN = 0.019、ω= 0.046)。如果SARS-CoV-2是从马来穿山甲冠状病毒重组而来,需经19.8-55.4年。所以该研究结论不支持SARS-CoV-2由穿山甲冠状病毒近期重组而来的观点。
总的来说,本研究通过103个SARS-CoV-2的基因组分析,首次系统阐述了病毒在人群传播过程中的演化动态。依据第8782位和28144位突变将病毒分为L型和S型,且在病毒暴发早期以L型为主导,进行人为干预如医疗和隔离措施的加强后,选择压力的变化使从L型为主导变为S型主导,这一持续发展中的类型可能需要医疗机构的重点关注。此外,对于SARS-CoV-2的起源,本研究不支持由穿山甲冠状病毒近期重组而来的观点,而更倾向于是趋同演化的作用。
研究人员特别强调,103个病毒株基因组数据量较少,后续工作需要扩大样本量,以验证这些结论或推测。此外,病毒分型的依据突变处在orf1ab中,它编码病毒基因组所必需的复制/转录酶,对感染机制可能也有影响。所以病毒不同亚型与其致病性的关系还不清楚,可以通过把基因组数据和病例结合起来分析,获得毒力/致病性的更多证据。但由于种种原因,研究者无法得到患者资料,也无法得到更多基因组数据。
此外,作者还强调,新冠病毒不是新近发生变异而分化成L和S型,而是这两个突变型在病毒暴发的早期可能就已经存在,是现在的隔离和医疗措施把可能更有害的L型频率给降低了。这一结果说明当前防疫和治疗的策略及措施是完全正确、并富有成效的。当然,他们的结果是建立在非常少的样本量,并且主要是基于分子演化的结果。更全面的病毒演化信息还需要更多专家们用更多样品结合临床数据来验证。
值得注意的是,与本篇研究相同,中国疾控中心同日在bioRxiv的研究也依据8782位点和28144位点的不同突变将SARS-CoV-2毒株分为两种类型[4],两篇研究相互印证。
截至3月4日,GISAID中目前已上传158株SARS-CoV-2基因组序列,但中国大陆只有2月9日之前的毒株,且大都集中于1月29日之前,1月29日之后只有4例,想要获得更多的病毒演化相关的信息需要结合近期鉴定的病毒基因组序列进行分析。笔者衷心希望研究者们在科学研究上能够做到资源共享,共同推动科学发展。
再强调一遍,共享新冠病毒基因组序列在当前十分重要,这个时候有资源的研究团队千万不要藏着掖着!!!
参考文献
[1] Kang M, et al. Evidence and characteristics of human-to-human transmission of SARS-CoV-2. medRxiv. Posted February 17, 2020.
[2] Yu W, et al. Decoding evolution and transmissions of novel pneumonia coronavirus using the whole genomic data. Chinaxiv. Posted February 21, 2020.
[3]
[4] Wu A, et al. Mutations, Recombination and Insertion in the Evolution of 2019-nCoV. bioRxiv. Posted March 02, 2020.
[5] Grubaugh ND, et al. We shouldn’t worry when a virus mutates during disease outbreaks. Nat Microbiol (2020).
本文经授权转载自微信公众号“BioArt”,由两篇来自“BioArt”的文章整合而成。原文链接:;原文链接:。
话题:

0
推荐
财新博客版权声明:财新博客所发布文章及图片之版权属博主本人及/或相关权利人所有,未经博主及/或相关权利人单独授权,任何网站、平面媒体不得予以转载。财新网对相关媒体的网站信息内容转载授权并不包括财新博客的文章及图片。博客文章均为作者个人观点,不代表财新网的立场和观点。
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}