阅读:0
听报道
因为单颗粒冷冻电镜厚积薄发的技术突破,结构生物学在2013年底经历了一场“分辨率革命 (resolution revolution) ”,从此几家欢喜几家愁。欢喜的是,之前许多让科学家们束手无策的生物大分子们纷纷被揭开了神秘面纱;愁的是,作为研究者,为伊消得人憔悴、蓦然回首却在灯火阑珊处的乐趣少了许多。
但是科研永无止境。过去十几年,每当有人问我结构生物学的未来,我的答案从未改变:“实时观察细胞内部近原子分辨率的结构”。目前还没有技术可以达到这一时空分辨率,而最接近的就是cryo-ET (冷冻电镜断层成像)。可以预见,这个技术的不断发展终将带来另一场分辨率革命,其意义辐射生命科学许多分支。
Cryo-ET仍旧处于蓬勃发展的阶段,在我赴普林斯顿任教前的几个月,成功帮清华引进了年轻的Cryo-ET专家李赛博士。过去三年,我也一直在努力帮普林斯顿大学招聘cryo-ET方面的人才,现在却依然是进行时,因为这个领域的人才确实是凤毛麟角。《返朴》邀请李赛博士撰稿,简要介绍cryo-ET的原理和发展,以飨读者。
——颜宁
撰文 | 李赛(清华大学结构生物学高精尖创新中心研究员)、徐家璐(李赛实验室博士研究生)
病毒,是直径100 nm左右的超大分子复合物。它的尺寸恰好在电子显微镜的透射范围内,为了探明病毒的结构,需要不断优化电子显微镜的功能和电镜数据的处理方法。可以说,现代冷冻电镜方法学是与结构病毒学手牵手一起发展的。
冷冻电子显微镜——“眼见为实”的魅力
无论是列文虎克等科学家通过光学显微镜将细胞的结构展现在世人的面前,还是沃森和克里克发现DNA双螺旋结构,揭示基因遗传的分子机制,都向我们证明了科学研究中“眼见为实”的魅力。生命科学所研究的大多数情境,都发生在极为微小的细胞中。一个细胞的尺度可能比一根头发丝的十分之一还小,更别说组成细胞的蛋白质、脂质、核酸等等。因此,想要看清楚蛋白质等分子的结构,依靠传统的光学显微镜是无法做到的。
随着20世纪物理学的不断发展,科学家发现电子也是一种波。当电子束照射到某一个物体上时,电子会像光子一样携带物体的形状信息,并继续向前传播。又由于电子在磁场下会发生偏转,所以我们可以用磁场做出像放大镜一样的透镜系统。有了这两方面的理论准备,电子显微镜应运而生。由于电子的波长远小于光子的波长,电子显微镜的理论分辨率极限也远超光学显微镜的分辨率。在材料领域中,电子显微镜甚至可以观测到原子之间的排布。
尽管电子显微镜的放大能力无比强大,但并不是只要将细胞放在显微镜下就能看清楚里面的细节。由于电子很容易受空气中的灰尘或者空气分子本身的影响,电子显微镜的内腔需要维持在近真空下。而在真空中,细胞里面的水分子会瞬间沸腾气化(液体的沸点会随着气压的下降而下降),细胞也将随之烟消云散。所以,我们需要首先将细胞或者其他生物样本在极低的温度下瞬间冷冻,锁住它们在那一刻的状态,然后才能在冷冻电镜下进行观察。这也是冷冻电子显微镜成像(cryo-electron microscopy,简称cryo-EM)中“冷冻”(cryo-)两个字的由来。
除了需要冷冻以外,生物样品在冷冻电镜领域的应用还受到另外一个重要限制,那就是样品并不能经受过多的电子辐照。过多的电子辐照会破坏蛋白结构,给冷冻状态下的样品造成局部受热并引发膨胀,甚至可能会融化玻璃态的冰。因此,我们用冷冻电镜观测生物样品时,只能使用较低的电子剂量。但这就好比在漆黑的夜晚,不使用闪光灯就给人拍照,获得的照片将是模糊的。与之类似,使用冷冻电镜对生物样品进行单次拍照,信噪比也非常低。当然,科学家也有应对之道:首先纯化我们感兴趣的蛋白质,然后把溶解了成千上万个蛋白质分子的溶液极速冷冻后放在电镜里拍照。这时就可以获得几十万颗这种蛋白质的照片,并且每个蛋白质的方向各不相同,相当于对同一个蛋白质拍了几十万张不同的投影照片。将这些照片的信号叠加在一起,我们就能够提高信噪比,获得这个蛋白质的高清三维结构。这种方法也就是冷冻电镜单颗粒成像技术SPA(Single particle analysis)。
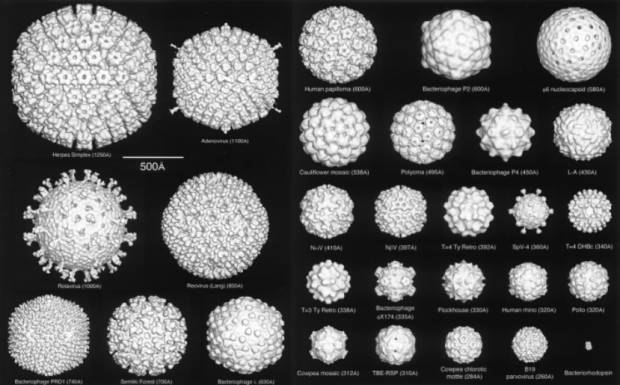
图1. 使用冷冻电镜解析的正二十面体型病毒的结构比较[1]。
断层成像:与其他结构生物学手段互补
随着直接探测电子相机(DDD camera)的出现和新算法的开发,冷冻电镜在2013年迎来一场分辨率革命(resolution revolusion),并逐渐成为结构生物学的主要研究方法。七年过去了,冷冻电镜单颗粒技术解析的高分辨率蛋白质结构如雨后春笋般显现,极大地丰富了人们对蛋白质微观结构与功能的认知。
与日渐成熟的冷冻电镜单颗粒技术一道,冷冻电镜断层成像技术cryo-ET(cryo-electron tomography)和子断层图像平均法STA(sub-tomogram average)也正在悄然崛起,将结构生物学引导向解析柔性超大分子复合物结构、细胞原位蛋白结构等方向,应用范围覆盖蛋白复合物、病毒、细菌、细胞甚至组织,分辨率最高已达3Å [2],是名副其实的跨尺度高分辨显微成像利器。
cryo-ET被应用在结构生物学中,可以回答很多独一无二的生物学问题,在笔者看来最吸引人的有四个:1)它带来丰富的、纳米级分辨率的生物背景 (context) 。比如生物大分子在原生环境中的拷贝数、分布规律、与其他分子的互作;2)它展示天然原位的生物结构;3)它的三维原始数据特点为解析柔性超大分子复合物带来可能;4)结合光电联合、聚焦离子束减薄等技术,它可以实现跨尺度高分辨成像,甚至可以从生物组织中获得纳米级分辨率三维信息。实际上,目前结构生物学领域积攒了太多只有cryo-ET才能回答的问题。由于应用前景广阔,可拓展空间巨大,cryo-ET被誉为结构生物学的未来技术方向。
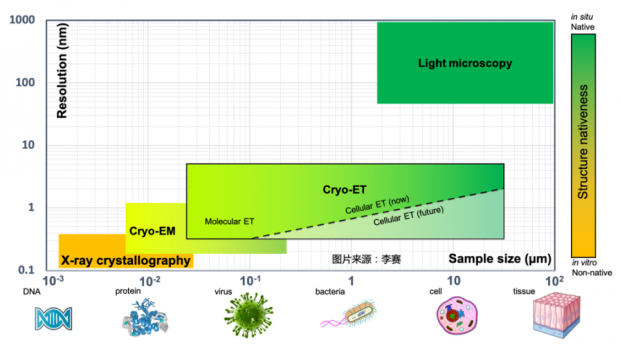
图2. cryo-ET是一种高分辨、跨尺度的原位显微成像方法,应用面非常广阔(图:李赛)
那么cryo-ET的原理是什么呢?
首先,cryo-ET的“T”和在医院里做的CT(computerized tomography,意为“电子计算机断层扫描”)的“T”是同一事物,都是“断层”的意思,二者的三维成像原理也很类似。在医院做CT检查,扫描仪器会绕着身体旋转并进行拍摄。而对于cryo-ET来说,需要旋转样品而不是电镜。通过旋转样品台,我们可以对样品进行不同角度的拍摄,一般是在-60°到+60°的范围内。
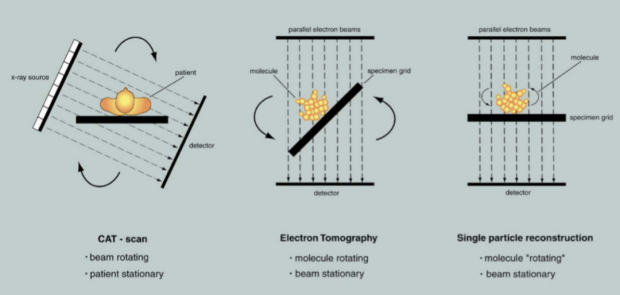
图3. CT、ET、EM的比较(图片来自Joachim Frank书籍)
从-60°到正60°,每隔3°对样品拍一张照,共可得到41张平面照片。这41 张照片足以构建样品的三维信息吗?从二维投影到三维图像的重构原理是什么?分辨率的限制是什么?这些关于cryo-ET的基础的思考诱发了几个重要理论的建立,它们奠定了cryo-ET技术的理论基础,是后人细化技术革新的理论坚石。
01 中心截面定理
中心截面定理是指,一个物体在某一方向上投影的傅里叶变换,等于该物体三维傅里叶变换中过中心的与投影方向垂直的截面。
根据中心截面定理,我们拍摄41张不同角度的照片,对这些照片做傅里叶变换,就能够得到这个物体三维傅里叶空间里41张截面的信息。我们再对这个被信息填充的三维傅里叶空间做逆变换,就能够获得该图像的三维形状。因而,我们可以通过cryo-ET技术获得细胞或者蛋白质的三维结构。
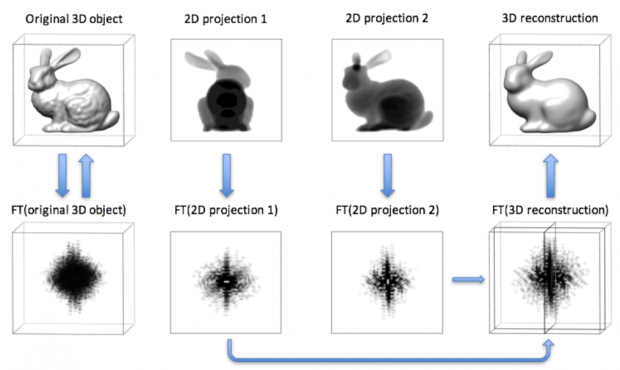
图4. 中心截面定理的示意图,三维的兔子被投影成二维照片后做傅里叶变换,与三维兔子直接做傅里叶变换后垂直投影方向的截面是一样的。(图片来自于牛津大学cryo-ET workshop课件)
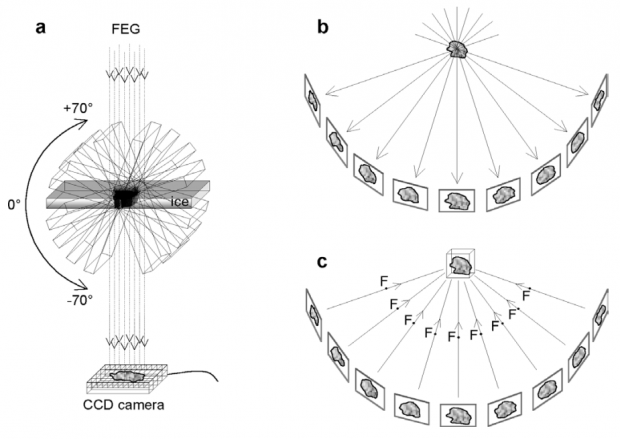
图5. cryo-ET成像示意图。(a)FEG表示电子枪,用于发射电子。CCD camera相机用于采集照片。光路下的样品被一边旋转一边拍摄;(b)表示对同一个物体进行了不同角度的拍摄;(c)代表通过这些不同角度的照片重构出物体原本的结构(Gruenewald et al., 1993)。
02 克劳瑟准则
细心的读者朋友也许发现了,41张照片远远不足以填充连续的三维傅里叶空间,只能够离散地对其做出不完整的填充。并且由于低电子剂量拍照的原因,每一个角度的照片都有着很高的噪音。其实,在cryo-ET技术发展的早期,科学家们已经对cryo-ET成像的潜力进行了物理与数学上的分析。剑桥分子生物学实验室(MRC-LMB)的结构病毒学家Tony Crowther等人在1969年分析了对于一定尺寸的物体,至少需要多少不同角度照片才能将其结构重构到一定分辨率,并提出了一项准则:克劳瑟准则 Crowther criterion [3]。
克劳瑟准则的公示表达是:N=πD/d。其中,D和d分别代表了样品的尺寸以及想要达到的分辨率,N是想要获得目标分辨率至少需要拍摄的倾转照片数量。例如,当样品是直径100nm的病毒,而我们想要获得的分辨率是10nm的话,那么我们所需要拍摄的不同角度数大约是31张。当然,克劳瑟准则的推导需要很多前提条件,但它提出的概念仍具有极强的指导意义:即使电镜拍摄的不同角度照片数量有限,我们仍可以获得纳米分辨率级别的三维结构信息。这表明冷冻电镜断层成像这一技术是具有研发和应用的前景的。
03 电子剂量分配定理
1976年,位于德国慕尼黑附近的马克斯普朗克生物化学所的Walter Hoppe (2017年诺奖得主Joachim Frank的博士导师) 又提出了关于电子剂量分配的定理Dose fractionation theorem[4]:假设使用同样的电子剂量拍照,把这些剂量平均分配给多个角度的照片(假设可以完美地对齐这些照片),那么相比于用该剂量一次性给单个角度拍照,从这些倾转照片获得的三维信息将更好。
简单说来,这意味着尽管cryo-ET的每个单张照片可能信噪比较低,但重构成三维图像后,信号可以加强,并且理论上信号质量可以超过单次高剂量投影相片。
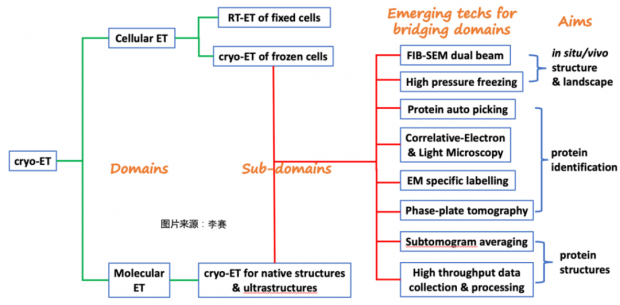
图6. 按照解决生物问题的不同,ET技术方向有多种层次的细分(图:李赛)
有了这些理论上可行性的分析和铺垫,cryo-ET技术在后来40多年的发展中被应用到蛋白复合物、病毒、细菌、细胞甚至组织等跨尺度高分辨成像中,在技术上不断细分并不断完善。
STA:原子级别的分辨率
了解单颗粒技术(SPA)的读者朋友也许会疑问,在使用单颗粒技术解析蛋白质结构时,往往需要对几十万颗同种蛋白质进行拍摄,才能够填补傅里叶空间,获得高分辨率的结构。那么cryo-ET中有类似的方法吗?答案是肯定的。
在cryo-ET收集的数据中,可能也会出现高度重复的同一种蛋白或者更大的复合体。例如新冠病毒原位全病毒结构[5],每套cryo-ET数据都包含了10多颗完整的新冠病毒,平均每颗新冠病毒的表面分布着30个刺突蛋白。既然这些刺突蛋白是同一种蛋白质,并且大量重复出现,就可以将其所在的子断层图像(sub-tomogram)裁剪出来,并旋转到同一个方向,把所有的该蛋白质的信号叠加起来,这样就可以获得一个高信号、低噪音的蛋白质结构,这也就是STA的原理。
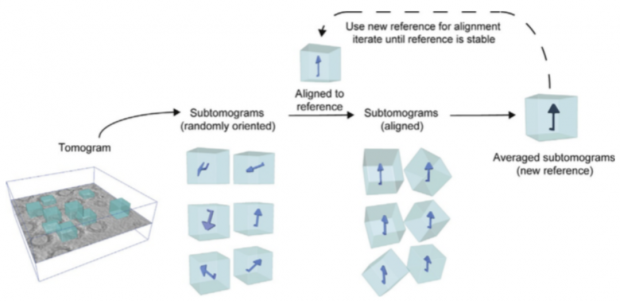
图7. 子断层平均法的示意图。Cryo-ET获得的断层成像中,相同蛋白所在的子断层被裁剪出来,并对齐到同一方向。这时,将所有子断层的信号进行叠加就能获得高信噪比的三维重构[6]。
由于刺突蛋白在原始三维数据中的准确坐标已经确定,因此在解出蛋白结构后,可以将其投射回去。通过这种方法,研究者从2300颗新冠病毒的cryo-ET数据中计算出最高达7.8Å分辨率的刺突结构,以及13Å的核糖核蛋白复合物(RNP)结构,并投射重构出一个具有代表性的新冠病毒全病毒结构。

图8. 新冠病毒断层图像截面,叠加在原始数据上的使用STA重构的完整病毒结构,以及刺突蛋白最高达7.8Å分辨率的结构[5]
读到这里,读者朋友们可能会感觉:子断层平均法STA与单颗粒技术SPA的原理很像,都是将同一种蛋白质的信号大量叠加在一起来提升信噪比,事实上也确实如此。当然,子断层平均法目前的分辨率还比不过单颗粒技术,原因有很多,可能是拍照效率不够高、拍照过程中样品抖动、生物样品受到电子辐照损伤等等。
单颗粒技术每次只需要拍一张照片,这个过程可能只需要半分钟,而使用断层成像时,则要倾转样品,又要保持倾转的过程中样品不被挪出拍照视野,所以一套cryo-ET的数据采集相对费时,大约需要半小时。这样一来,使用cryo-ET方法很难获得像单颗粒方法一样多的数据。数量上不去,最终的分辨率自然受到限制。
另外,电镜的机械装置在倾转样品时,难免会导致样品的前后位置对应不起来。这就好比在拍CT时,尽管医生交代了需要保持静止,患者却左右晃动。这会造成最终重构出的结构像拍摄奔跑的运动员一样带着糊影。最后,过多电子的照射会使生物样品所在的冰层发生形变,这和样品的抖动一样,也会造成分辨率的下降。
以上这些限制cryo-ET分辨率的因素,造成了目前大多数cryo-ET和STA应用所获得的分辨率一直停留在4Å以外。在2015年,被广泛使用的单颗粒重构软件Relion的研究团队在Structure杂志上发文,把Relion的算法应用到了STA上[7]。作者在论文中明确指出,电子辐照产生的样品移动是限制STA分辨率的重要因素。具体来说,不同角度之间的样品移动只能通过金颗粒进行校正,其准确性是不足的。
但科学家们一直在不断努力打破分辨率的极限。2018年,牛津大学章佩君团队开发了emClarity软件,首次实现了以样品颗粒为基准来校准不同角度之间样品的移动,并通过循环来实现校准,获得了3.1Å的核糖体(体外提纯样品)STA结构[8]。
今年,位于德国哥廷根的马克斯普朗克生物物理化学研究所Patrick Cramer团队的Dimitry Tegunov等人开发了M软件[10]。结合使用M及他们之前开发的Wrap软件[9],能对电子产生的样品移动及冰层形变进行更为精确的校准。这一算法成功地将细胞切片的cryo-ET数据中的核糖体解析到了3.7Å的分辨率,并清晰地展示了其中的抗生素电子密度。这一令人震惊的结果预示着cryo-ET和STA的高分辨时代也许即将到来。在不久的将来,也许我们可以将原位的蛋白结构广泛地解析到近原子分辨率,使用cryo-ET来获得蛋白质的原子模型将成为可能。而在这背后作为支撑的,则是越来越稳定高效的电镜硬件,性能越来越高的计算机,以及考虑越来越精细的算法。
囊膜病毒的“照妖镜”
在冷冻电镜结构病毒学的发展过程中,加州大学圣地亚哥分校的Timothy Baker和牛津粒子成像中心的首届主任Stephen Fuller起着奠基者的作用。Baker坚持研究以正二十面体型为主的无囊膜病毒 (nonenveloped virus),而Fuller则选择了对人类健康威胁更大的囊膜病毒 (enveloped virus)。无囊膜病毒没有囊膜,直接由蛋白质外壳包裹,其范畴较广,从动物病毒、噬菌体、到水生病毒等都有。囊膜病毒的外围则有脂质双层膜包裹,侵扰人类的天花、乙肝、艾滋、狂犬、流感、寨卡等病毒,均是囊膜病毒。
虽然都是病毒,但针对两者的研究手段有很大的区别。无囊膜病毒大多数就像一个模子刻出来的,典型的结构是正二十面体组装(图1),其结构解析主要使用冷冻电镜成像技术cryo-EM及单颗粒重构方法SPA。而绝大多数囊膜病毒组装松散,柔性极大,高分辨率重构整颗囊膜病毒的唯一方法,就是采用冷冻电镜断层成像技术cryo-ET和子断层图像平均法STA。
为了解析囊膜病毒结构,Fuller从此迈入cryo-ET这个在当时异常年轻的领域。
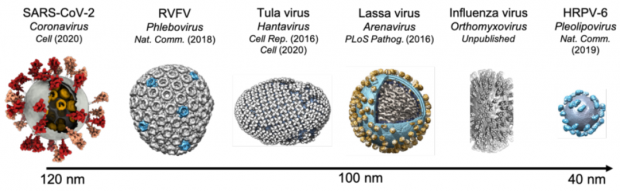
图9. 使用冷冻电镜断层成像和子断层图像平均法解析的囊膜病毒比较。自左至右:新冠病毒、裂谷热病毒、汉坦病毒、拉沙病毒、流感病毒、盐生多形型病毒(来源:李赛实验室及李赛项目图片)
2000年,Fuller辞去海德堡欧洲分子生物学实验室(EMBL Heidelberg)主任一职,搬到有多年结构病毒学传统的牛津大学结构生物学部,并在那里与学部主任David Stuart(饶子和院士的博士后导师)联手建立了牛津粒子成像中心,并任第一届主任。他主持建成了集成高端冷冻电镜设施的BSL-3(生物安全三级)实验室,并在那里开展艾滋病毒HIV的成像及结构工作。至今,全世界同一级别、功能类似的实验室仅有两家,另一家在美国德克萨斯大学医学院 (UTMB),主要从事病原微生物的鉴定、病原-宿主相互作用及结构解析等方面的研究。
肆虐全球的新冠病毒(SARS-CoV-2)亦属于囊膜病毒。2020年,在新冠病毒基因序列公布之后的3个月内,重组表达的刺突蛋白、核蛋白的局部结构已经被解析出来了。然而,病毒的完整结构、结构蛋白在病毒上的原位全长结构、分布规律、拷贝数及与其他结构蛋白的互相关系仍是谜团。
如果能尽快将病毒的形象真实、完整、清晰地呈现给世界,也许就能让更多人对病毒有直观认识,对疫情防治重视起来。今年八九月间,Nature [11]、Science [12]、Cell [5]杂志分别报道了英国剑桥、德国马普所、清华大学的研究人员解析新冠病毒原位结构的研究。众多网友纷纷评论:“病毒长得很瘆人”“毛骨悚然”“一看就不好惹”“为新冠病毒张贴了高清的通缉照”……可见完整真实的病毒形象的确带来了视觉冲击,起到了警示作用。
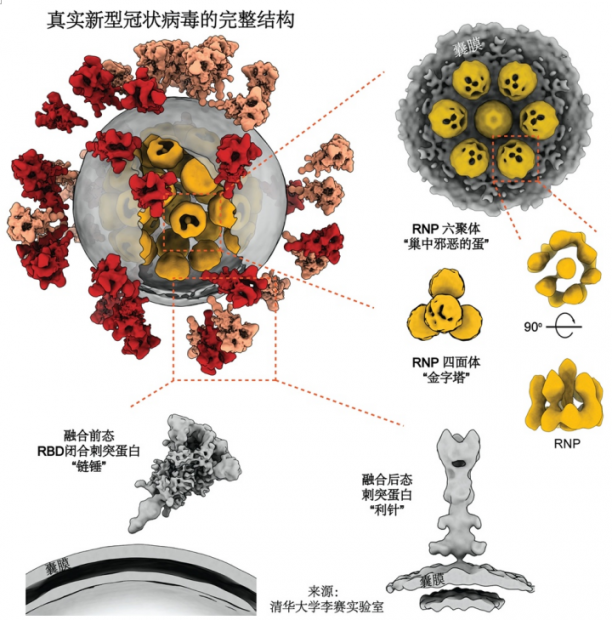
图10. 真实的新冠病毒全病毒结构[5]。
冠状病毒的“皇冠”特征,来源于病毒表面凸起的刺突蛋白(spike protein)。它就像一把“钥匙”,让新冠病毒识别出细胞表面的受体,并与之结合,介导膜融合,最终侵入细胞。目前大多数疫苗和抗体的研发都聚焦于刺突蛋白。在全病毒结构解析的过程中,研究人员发现,相比于其他囊膜病毒,新冠病毒的刺突蛋白有三个独特的地方:
(1)在病毒表面分布随机,且拷贝数较少(平均约30个),仅为拉沙病毒及流感病毒的1/5-1/10;
(2)靠近病毒囊膜的茎部区柔性较大,使得刺突蛋白在病毒表面能够近乎自由地旋转,甚至可能游走,就像古代的武器“链锤”一样。因此新冠病毒在侵染细胞时,能够自由调整刺突蛋白的方位,方便和单个、甚至多个受体结合,这可能是新冠病毒高传染性的原因之一;
(3)刺突蛋白在病毒表面可呈现两种状态:其中97%处于“融合前状态”(prefusion),3%处于“融合后状态”(postfusion),只有后者才能和ACE2受体结合。一般而言,刺突蛋白只有受到pH值变化、受体结合等因子触发才会向融合后状态转变,而新冠病毒带有疑似“自发”的融合后状态。这种状态的刺突蛋白已经缺失了其S1亚基,剩下的S2亚基已经发生较大的形变,也许能“欺骗性”地诱导出一些抗体,但这种抗体可能无法中和正常的病毒。
除了解析病毒表面的刺突蛋白结构,清华大学的研究人员还在仔细查看新冠病毒的断层图像过程中,发现病毒体内塞满了空心珠子一样的东西,而且这些珠子似乎有着排列规则。在贴近病毒囊膜上下表面的地方,有时可以看见这些珠子排列成清晰的六边形。经验表明,这些珠子可能就是核糖核蛋白复合物RNP,而且它们是有多个层次结构的。
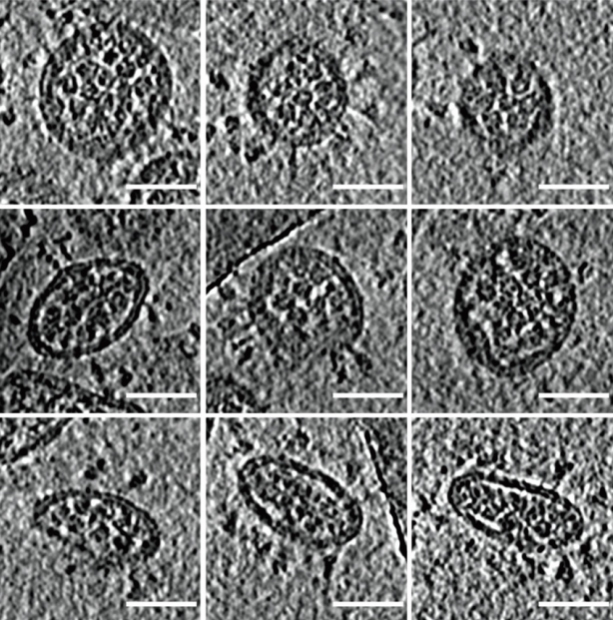
图11. 新冠病毒体内有很多珠子一样的物质,而且它们隐约有排列规律。标尺为50纳米(图:李赛)
经过挑选和分析,研究人员展示出,新冠病毒腔内的RNP像串珠一样将RNA组织在一起,并在病毒体内呈现六聚“鸟巢”型和正四面体“金字塔”型两种局部排列,有序地收纳了长长的RNA链条,还增加了病毒在复杂环境中经受物理挑战的能力。这可能是世界范围内首次“看清”正义单链RNA病毒的内部结构。
以上三篇解析新冠病毒全病毒结构的文章实际都得益于Stephen Fuller早年种下的大树。Nature一文的通讯作者John Briggs是Fuller的博士生;Science一文的第一作者Beata Turnova是John Briggs的博士生;Cell一文的通讯作者李赛在牛津粒子成像中心做了5年博后/副研,可算是Fuller的第三代弟子。令人痛惜的是,Fuller因健康原因56岁便退休,60岁时便英年早逝(2014年)了。他虽未能亲眼目睹冷冻电镜在后来突飞猛进的技术革命,但他创始的囊膜病毒结构学及参与奠基的cryo-ET技术现在已是遍地开花。愿以此文纪念Fuller教授。

图12.(左)牛津大学粒子成像中心创始人之一Stephen Fuller教授及其团队的自画像;(右)该中心BSL-3级冷冻电镜实验室内景。(来源:李赛)
参考文献
[1] T. S. Baker, N. H. Olson,S. D. Fuller, Adding the third dimension to virus life cycles:Three-dimensional reconstruction of icosahedral viruses from cryo-electronmicrographs. Microbiol Mol Biol R 63, 862-+ (1999).
[2] D.Tegunov, L. Xue, C. Dienemann, P. Cramer, J. Mahamid, Multi-particle cryo-EMrefinement with M; visualizes ribosome-antibiotic complex at 3.7 Å insidecells. bioRxiv10.1101/2020.06.05.136341, 2020.2006.2005.136341 (2020).
[3] D.J. D. R. A. Crowther, A. Klug, The reconstruction of a three-dimensionalstructure from projections and its application to electron microscopy. Proceedings of the royal society A (1970).
[4] R.Hegerl, W. Hoppe, Influence of electron noise on three-dimensional imagereconstruction. Zeitschrift fürNaturforschung A 31, 1717-1721(1976).
[5] H.Yao et al., Molecular architecture ofthe SARS-CoV-2 virus. (2020).
[6] W.Wan, J. A. Briggs, Cryo-Electron Tomography and Subtomogram Averaging. Methods Enzymol 579, 329-367 (2016).
[7] T.A. M. Bharat, C. J. Russo, J. Lowe, L. A. Passmore, S. H. W. Scheres, Advancesin Single-Particle Electron Cryomicroscopy Structure Determination applied toSub-tomogram Averaging. Structure 23, 1743-1753 (2015).
[8] B.A. Himes, P. Zhang, emClarity: software for high-resolution cryo-electrontomography and subtomogram averaging. NatMethods 15, 955-961 (2018).
[9] D.Tegunov, P. Cramer, Real-time cryo-electron microscopy data preprocessing withWarp. Nature methods 16, 1146-1152 (2019).
[10] D.Tegunov, L. Xue, C. Dienemann, P. Cramer, J. Mahamid, Multi-particle cryo-EMrefinement with M visualizes ribosome-antibiotic complex at 3.7 Å inside cells.bioRxiv 10.1101/2020.06.05.136341,2020.2006.2005.136341 (2020).
[11] et al., Structures and distributionsof SARS-CoV-2 spike proteins on intact virions. Nature 10.1038/s41586-020-2665-2 (2020).
[12] B.Turonova et al., In situ structuralanalysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 10.1126/science.abd5223 (2020).
作者介绍

李赛从事冷冻电镜断层成像技术开发及囊膜病毒的结构研究。2009-2012年,他在德国哥廷根大学物理学院从事流感病毒组装机制研究,并获得博士学位;2012-2018年,在牛津大学结构生物学部的BSL-3级实验室开发及使用cryo-ET方法,并研究了拉沙病毒、裂谷热病毒、汉坦病毒及嗜盐古菌多形病毒的高分辨结构;2018年入职清华大学生命学院并建立独立实验室,开展了冠状病毒、艾滋病毒等研究。在过去12年的囊膜病毒研究中,他较为系统地研究了新发致病型囊膜病毒的组装,及通过膜融合去组装的原位结构与机制,并希望找到病毒的弱点。为解答这些问题,他一直深耕于高分辨率 cryo-ET方法的开发及应用。他的工作发表在Cell、Cell Research、Nature Communications、Cell Reports、PLoS Pathogens等刊物上。
话题:

0
推荐
财新博客版权声明:财新博客所发布文章及图片之版权属博主本人及/或相关权利人所有,未经博主及/或相关权利人单独授权,任何网站、平面媒体不得予以转载。财新网对相关媒体的网站信息内容转载授权并不包括财新博客的文章及图片。博客文章均为作者个人观点,不代表财新网的立场和观点。
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}