阅读:0
听报道
去年年末,谷歌公司旗下DeepMind团队研发的AlphaFold2人工智能系统在国际蛋白质结构预测竞赛(CASP)上取得惊人的准确度,多数预测模型与实验测得的蛋白质结构模型高度一致,引起举世瞩目。《返朴》曾发表多位相关领域科学家的评论(回顾:《颜宁等点评:AI精准预测蛋白质结构,结构生物学何去何从?》)。
时隔半年,7月16日,DeepMind在《自然》杂志上发文,公开了进一步优化的AlphaFold2源代码并详细描述了其设计框架和训练方法。几天后,DeepMind又发布了由AlphaFold预测的蛋白结构数据库,免费提供给全球科研人员开发使用。对此,《返朴》总编、结构生物学家颜宁提出了自己的见解。
最近这次AlphaFold带来的震撼其实不如第一次大,因为有了上一次的突破,现在这个结果基本是水到渠成。对比了一下钠离子通道的结构——预测的部分与电镜已经解析的部分吻合得还行,但没有任何homology model(同源模型)的部分,比如电镜结构里看不到的部分,在预测的结构里依然是一团无序的圈圈。
AlphaFold毫无疑问是对整个生物学的一场变革,而不局限在结构生物学领域。但它并不是结构生物学的终点,而是可以把结构作为起点去做更多的东西。生命在于运动——分子层面的运动是冷冻电镜技术革命之后结构生物学新的重点。以我的科研为例,我们的目标并不是要获得研究对象的折叠信息,而是要解析其处于不同工作状态的精准构象信息,以此来理解它的工作机理和致病机理。
短短几天,学界针对AlphaFold已经形成两派:做超级复合物的叹为观止,做膜蛋白的,比如我,感觉帮助不大,并且误差较大。这是为什么?
有人认为,是因为膜蛋白有脂水两相;有人认为,是因为膜蛋白本身解出来的可靠数据少,不够供AI训练;还有评论说,是因为膜蛋白静态简单,动态复杂;超级复合物则相反,而AlphaFold强在静态蛋白结构预测。
膜蛋白的折叠有着相对复杂的脂分子环境,一旦缺少可靠的模型,AlphaFold所预测的蛋白质折叠可能大体正确,但在具体构象上与实验数据有较大的差距。而决定膜蛋白复杂功能的,不是它相对简单的折叠模式,而是精细的构象变化。
AI最大的挑战,将会是在未来。关于分子的动态研究、动态预测、动态模拟等等,其实还有很多问题都值得解决。在上次AlphaFold横空出世的时候我就说过,希望AI下一步可以解决分子动力学模拟对于很多生物过程无能为力的问题。那就继续期待AlphaDynamics吧?
——颜宁
撰文 | 继省
最近一周以来,生物医药圈子被AlphaFold v2.0刷屏,每天打开微信朋友圈,都能看到至少一半的新消息与它有关。这款由谷歌旗下人工智能公司 DeepMind团队开发的蛋白三维结构预测程序,在2020年的蛋白结构盲测比赛CASP中一骑绝尘独领风骚;而这次研发团队把全部代码任性地开放,并把Uniprot*上大部分代表性蛋白序列都给预测了一遍。
*UniProt是一个免费使用的蛋白质序列与功能信息数据库。
与此同时,蛋白设计领域的大神David Baker课题组参考AlphaFold的思路,利用他们长期在蛋白质设计领域的优势推出了一款同类软件RosettaFold。后者虽然不如AlphaFold覆盖全面,但在一些代表性的蛋白结构预测上已经可以与之媲美,并且对计算资源的占用更少。
我偶尔也会八卦:是不是RosettaFold的良性竞争压力促成了AlphaFold的全面开源?总之,一时间结构生物学、人工智能、合成生物学还有生物医药投资等多个领域的研究者开始了热烈的讨论。
但热闹是他们的,我只关心我课题里的蛋白能不能利用这两个程序的某一个来凹个造型,哦不对,预测个结构。
对于绝大多数实验生物学从业者来说,我们在实验室中研究某个蛋白功能之后,常想进一步了解一下它们的空间结构,这样就能够更好地理解和诠释我们在实验中看到的一些现象,也可以针对空间结构去开发设计一些药物,来阻断蛋白的功能。——当然,这部分设计需要另外的专家。
AlphaFold把预测结果以数据库的形式公开在了网络上。对于只想薅羊毛的用户,不啻天外福音。于是我兴致勃勃地打开了它的预测结果查询网站()。
我一看,直接输入蛋白或者基因名就可以了,非常符合我这种不懂结构生物学和人工智能的选手。
好,先来一个试试。以前读博士时,经常研究一种叫beta-catenin的蛋白,这是一个在脊椎动物个体发育和癌症等多个生物学过程里都非常重要的蛋白。输入蛋白名称后,得到25条结果,对应人、大鼠、小鼠、斑马鱼等多个模式物种。这25条当然不是现在地球上已知叫beta-catenin的全部蛋白,但能有模式物种的结果,就很有代表性了。
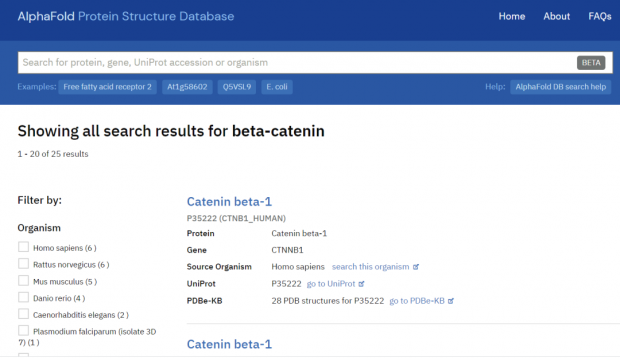
排在第一位的链接,是人的beta-catenin。就可以看到一个五颜六色的三维结构——不同颜色代表着AlphaFold预测的可靠性,深蓝色和浅蓝色代表AlphaFold对预测模型很有信心,而橙色和黄色代表他们也不太确信。可以看到,beta-catenin蛋白中间12个像弹簧一样的alpha螺旋,这类连在一起的alpha螺旋有个诨名叫Armadillo repeat,得名于动物犰狳(Armadillo,如下图)。

()
怎么样,这两家伙还挺像吧?
但是预测出来这个犰狳重复并不出奇——PDB数据库*现已发表数个人类beta-catenin结构,说明这段序列比较稳定,换言之就是供AlphaFold学习训练的知识比较充沛,它预测也比较容易。反倒是这段重复区域的前后两端(N端和C端)还各有近百个氨基酸,至今没有稳定的结构问世(当然,可能蛋白本身在这个区域就属于比较混沌无序的),而AlphaFold的模型里,在N端(氨基端)和C端(羧基端)也同样分数很低,并没有显著改善。
*PDB,全称Protein Data Bank,是目前最主要的收集蛋白质三维结构的数据库。
再试验一个我现在导师课题组里研究多年的膜蛋白TGFBR2,这是一个受体酪氨酸激酶,既是膜受体又是激酶,同样没有全长结构问世。同样的流程,找到人TGFBR2,打开结构后是下面的情形:这个蛋白明显出现了三个分数比较高的区域,包括靠近N端的配体结合区,中间的跨膜区,和C端的激酶区,从序列上看,和目前人们对这一蛋白的功能认识很吻合。而画面中橙黄色低分区域,也同样是PDB已有结构里缺失信息的部分。
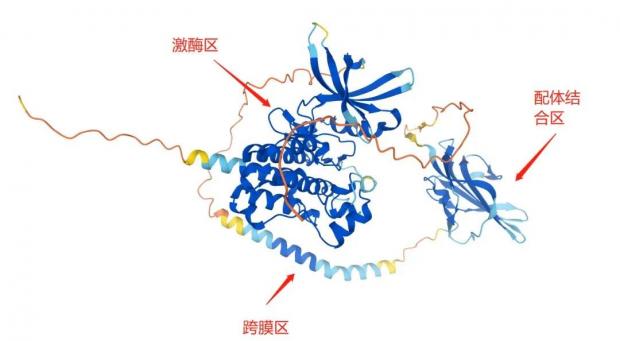
单从这两个例子看来,AlphaFold的确可以复现科学家实验得到的蛋白结构。尤其是这个激酶区,套用网上流行的俏皮话来说,不说非常相似吧,简直是一模一样。
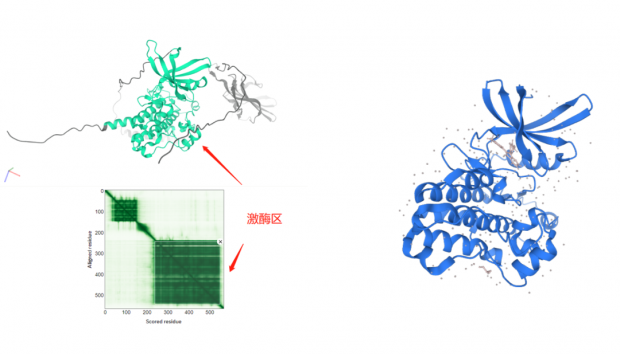
左图:AlphaFold预测的TGFBR2结构(中间为激酶区),下方的深绿色模块对应上方高亮的激酶区;右图:PDB数据库上的代表性TGFBR2激酶区结构,是实验得到的结构数据。
当然了,说一模一样太夸张,还是有细微差别的。最重要的一点,右侧实验得到的结构里有一个化学小分子结合到蛋白上,这是一种生理调控下的蛋白状态;蛋白在有无化合物结合、不同化合物结合的状态下都会呈现不同的细微变化,而正是这些变化才体现了蛋白质的多样而神奇的功能。在这一点上,AlphaFold给出了一个单一的最优解,是无法覆盖蛋白质的千姿百态的。
但即便如此,AlphaFold也是做到了此前人们无法想象和做到的事情。
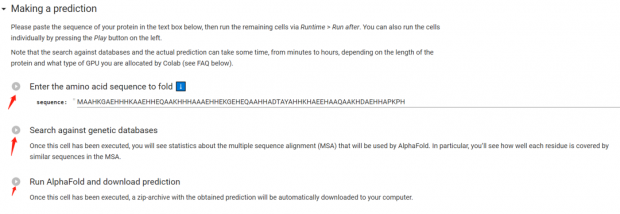
做了两个测试之后,自然就想到,如果随意给一段序列,能否轻松地得到一个结构模型呢?毕竟这才是AlphaFold最让人兴奋的地方。AlphaFold的正式运算需要的资源非常庞大,个人或者小团队如果家里没矿的话,想自己搭平台运算基本没戏;好在谷歌提供了一个免费的云端平台Colab(),可以运行精简版的AlphaFold。平台入口就在AlphaFold数据库的底端FAQ里。
AlphaFold Colab界面
这个云端平台解放了硬件,只要你准备好了要预测的氨基酸序列就可以了。当然运算方法比完整的AlphaFold要“简陋”一些。但聊胜于无,要啥自行车呢?
结构预测前的准备工作有三步:第一步连接平台,第二步安装第三方软件,第三步下载AlphaFold。连接平台需要登录(似乎只能用谷歌账号,不太确定国内的同行们能否使用),安装软件和下载AlphaFold只需点一下鼠标,各需2-3分钟。当真是哪里不会点哪里,So easy!
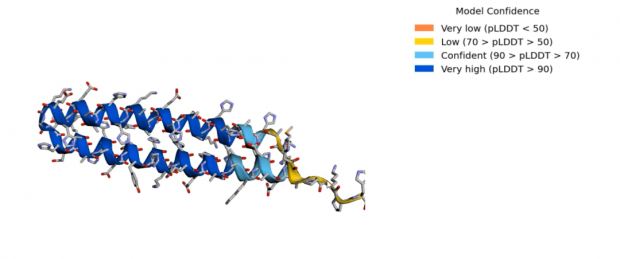
下面进入正式预测环节,我用的是系统输入框里自带的序列(长约70个氨基酸,经我查证这是某种细菌里的未命名蛋白)。同样是三步,第一步把冰箱门打开把序列输进去;第二步搜库,也就是看看已知结构里有没有序列和输入序列相似的;第三步给出一个最优解。晚上做到搜库时,系统显示要几十分钟,我就去睡觉了。结果早上醒来发现,搜是搜完了,但因为长时间没有动作,系统把我踢下来了,我又要重新开始。然而不管我是重新搜库,还是从头装软件,到了搜库这一步总是遇到同一个模块丢失的错误。凭我相当有限的编程知识,我重启了一下电脑,解决了图片。经过了约半小时的搜库和约15分钟的计算建模之后,我得到了一个很漂亮的回形针蛋白,从图中可以看到,可信度还是比较高的。
后来我咨询了一位懂编程的朋友,他告诉我可以把每一步的代码粘在一起,这样就不用手动一步一步地点击触发程序了。而且可以把Colab绑定谷歌云盘,这样就有更多的存储空间可以用了。这可能是懂编程的人们都知道的事情。要不咋说我是外行买家秀呢。
在AlphaFold搜库的半小时里,我还探索了一下RosettaFold。华盛顿大学David Baker实验室课题组搭建并公开的这个服务器每周都能吸引超过5000条计算任务,我用Colab上的示范序列来测试RosettaFold的预测效果,提交了任务之后发现前面还有3000个任务在排队,且等着吧。从运算时效上来看,还是AlphaFold Colab更快。有意尝鲜且手里有实验数据的朋友,不妨两个平台都试试。
前面我在AlphaFold数据库里检索的beta-catenin和TGFBR2两个蛋白,其实属于已知结构的蛋白了,Uniprot上有大量尚无PDB结构的蛋白条目,在AlphaFold上都有预测结果。比如AlphaFold数据库首页搜索框下方的示例Q8W3K0,这是一种存在于模式植物拟南芥里的蛋白,可能具有抗病作用。AlphaFold预测出来的结果惊艳到我了,蛋白好漂亮!难怪无数俊男美女投身到蛋白结构解析的大军中,这真的是门艺术。
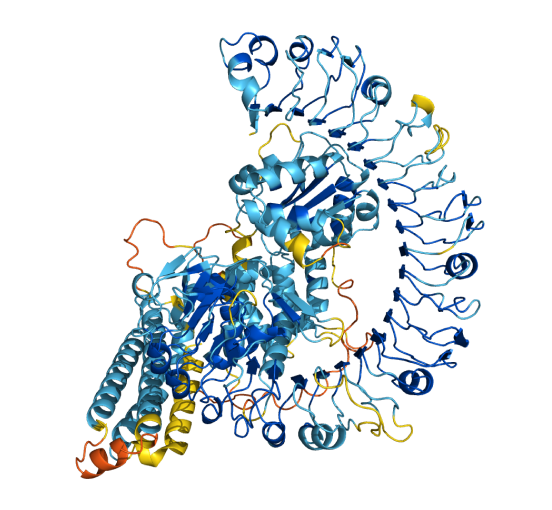
At1g58602蛋白预测结构(UniProt编号Q8W3K0)
以上就是我简单的体验经历。从我不成熟也不算专业的角度来看,AlphaFold对结构生物学家的冲击可能不大:一来,蛋白的无序区域(指不能形成稳定三维结构的区域)涉及氨基酸分子的自由运动,个中规律仍然需要结构生物学家去揭秘;二来,很多重要的生理病理性蛋白(如离子通道蛋白),在不同的活性状态下都会有很精妙的构象变化(conformational change),在细胞内外也会因为和各种各样的其他蛋白结合而呈现出千变万化的空间构象。以AlphaFold目前仅用蛋白本身序列作为输入信息的算法逻辑,至少还不能熟练应对上面提到的几种情形。
但AlphaFold的问世,本身就是一件打破常规超乎想象的创举,我们有理由期待这个工具会不断进化,持续给我们惊喜。如颜宁老师所说,也许在不久的将来,随着人们对蛋白质分子动力学知识的加深,引入分子动力学模拟的AlphaDynamics会横空出世,来预测出同一个蛋白的多个稳定构象。而对广义的实验生物学家来说,AlphaFold无疑提供了一个全新又容易上手的虚拟验证平台——设计一款融合蛋白,一个全新的小肽,或是给自己中意的蛋白制造几个氨基酸突变,拿AlphaFold的预测结果和实验结果相互佐证,会让本就刺激的工作更加有趣,也可以为AlphaFold未来的进化提供了更多真实数据。
未来可期!愿大家折叠快乐!
话题:

0
推荐
财新博客版权声明:财新博客所发布文章及图片之版权属博主本人及/或相关权利人所有,未经博主及/或相关权利人单独授权,任何网站、平面媒体不得予以转载。财新网对相关媒体的网站信息内容转载授权并不包括财新博客的文章及图片。博客文章均为作者个人观点,不代表财新网的立场和观点。
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}