蛋白质是一类重要的生命大分子,参与组成了机体所有的重要部分。它是生命的物质基础,也是生命活动的主要承担者,可以毫不夸张地说,没有蛋白质就没有生命。
人们针对蛋白质的研究从未间断过,但同为蛋白质,不同的种类所受的关注度却是天差地别。到目前为止,大多数蛋白质的研究都集中在有限的一些蛋白质上,结果是热门的蛋白质正在越来越被人们所熟知,冷门的蛋白质的生物学功能,人们仍然知之甚少。
长此以往,已知功能蛋白质和未表征蛋白质之间的巨大“鸿沟”便开始慢慢显现了。因此,重视对未表征蛋白质的研究,缩小不同种类蛋白质之间的认知差距,解决注释非均一性问题,可以说是箭在弦上,势在必行。
今年,Georg Kustatscher等六国科学家联合发起了一项名为“未充分研究的蛋白质计划”的调查,并分别在Nature Methods和Nature Biotechnology上发表了两篇重要文章,呼吁学界通过系统地关联未表征的蛋白质和已知功能的蛋白质,缩小两者之间的注释差距,从而为详细的机制研究奠定基础。

这项计划将如何开展?又将带给我们多少未知的惊喜?本文在不改变原文意思的基础上做了编译。希望更多的研究人员和科学家们可以响应这些呼吁,解密那些未知的未知和“暗物质”蛋白质,扩大人类对生物大分子认知的边界。
编译 | 纪十
责编 | 李晓韦
1、蛋白质研究现状
科学家们对于不同蛋白质的关注天差地别,人类蛋白质组中最受欢迎的蛋白质是p53,平均而言,每天有两篇以p53为研究对象的文献发表。
与此同时,数以千计其他人类蛋白质的生物学功能尚未得到研究。在人类蛋白质组功能表征研究中,95%的发表作品都在关注5000种已得到充分研究的人类蛋白质。人类基因组的测序被视作是减少这种偏见的关键,但即使在基因组序列公开十年后,75%的发表作品仍然只关注基因组绘制之前已被研究的基因。
事实上,自人类基因组序列发布以来,这种注释的非均一性(annotation inequality)几乎翻了一番。注释非均一性阻碍了生物医学的进步,基因疾病之间的关联机制研究通常集中在广为人知的蛋白质上(图1),与此同时,许多与疾病相关的未表征的蛋白质没有经过功能研究。
例如,许多参与罕见疾病(加总起来并不罕见)的蛋白质的功能,我们对它们知之甚少。此外,神经发育障碍和癌症等常见疾病是由不同基因中许多罕见的遗传变异共同引起的。
值得注意的是,截至2015年,在对一个人类细胞系增殖至关重要的1,878个基因中,有330个(18%)仍未被表征。这种偏差延伸到了目前预计可做药物研究的约3,000种蛋白质:目前只有5-10%的潜在靶标蛋白质可作为获FDA(美国食品与药品监督管理局)批准药物的作用靶点。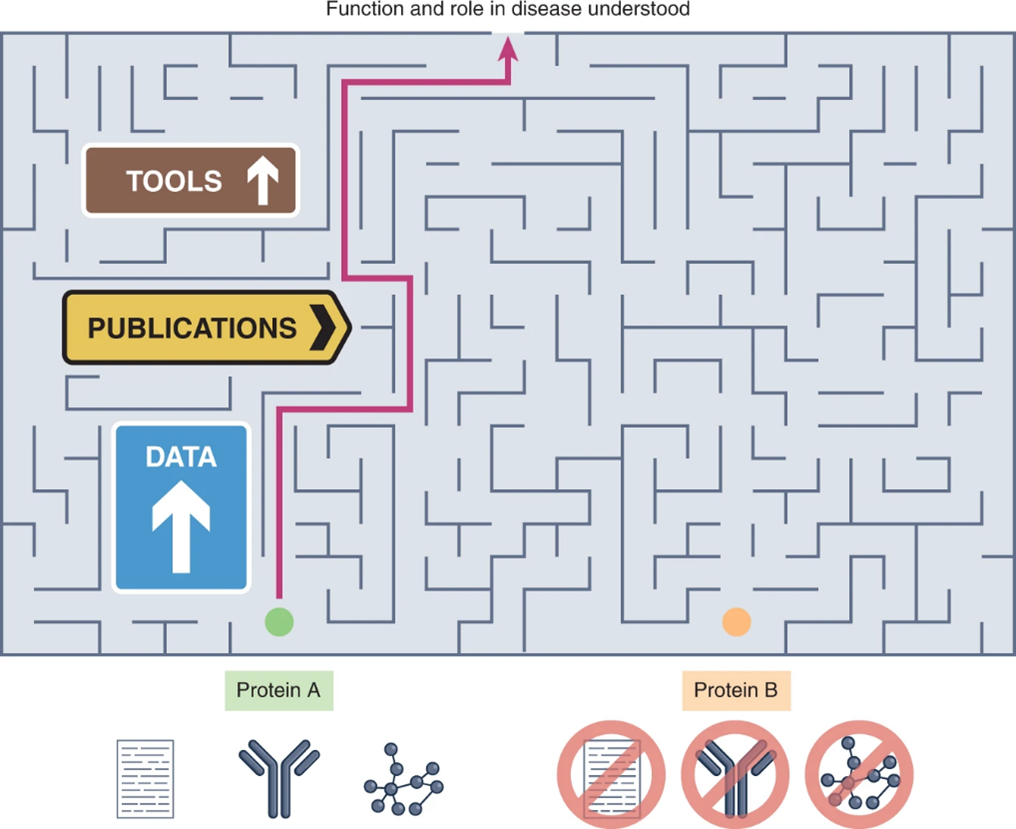
图1 蛋白质注释非均一性阻碍了生物医学的进步。以前的文献、数据和工具的数量决定了蛋白质研究问题是否容易提出和解决,这深化了注释偏差,也使得未充分研究蛋白质这一问题迟迟得不到解决。
2、蛋白质注释非均一性的起源
蛋白质注释偏差的原因是多方面的,有些是实际操作的原因,例如,抗体、质粒或精心打造的参考数据等实验工具有力地推动了人们选择研究那些已被充分研究的蛋白质。关于蛋白质的文献数量也与其基本的生物学和生化特性有关,例如蛋白质大小、丰度、疏水性及其基因对突变的敏感性。事实上,迄今为止,在19,733个人类蛋白质编码基因中,有1,899个(9.6%)缺乏蛋白质组学技术的可靠支持,其中一些可能构成基因组注释错误。
此外,未被充分研究的蛋白质的一个显著共同特征是分子量极小:SwissProt中注释最少的蛋白质中有40%小于15 kDa。此外,目前被人所知的未充分研究的“小蛋白质”库可能只是冰山一角,因为我们才刚刚开始发现“替代蛋白质”群,这些蛋白质的基因以前被认为是非编码区域。蛋白质注释非均一性的其他成因可能更反映了研究系统的偏见。
例如,人们通常仍会认为有众多研究的蛋白质在功能上更重要。科学家通常更愿意详细探索已经被研究过的问题,除了资金申请和同行评审系统需要规避风险外,投入大的研究领域还可以增加被引,增加在高影响力期刊发表文章的机会。支持现有范式而不是新思想的范式也减缓了整体科学进步。实验室研究的一些条件限制也是同样重要的原因。
矛盾的是,这些限制本意可能是希望通过实验室条件的标准化使研究产生更具可重复性的结果。例如,在标准的实验室培养条件下,约20%的酿酒酵母基因缺失会产生致死表型。然而,当培养空间扩大时,97%的基因对于在至少一个条件下的最佳生长至关重要。对于具有特殊细胞类型的多细胞生物体,这个问题更复杂;一些组织或细胞类型比其他组织或细胞类型的研究多得多。
最后,蛋白质注释偏差反映出人们更关注假设驱动而不是问题驱动的研究,而对非表征蛋白质的机制分子功能提出假设十分困难。哲学家弗朗西斯·培根(Francis Bacon)曾被认为是“科学方法之父”,他认为实验不应该由假设驱动,以规避观察者的偏见。因此有人提出,严格的数据驱动方法可以帮助减少蛋白质注释非均一性。
3、加速针对未充分研究蛋白质的药物发现
从药物发现的角度来看,那些提高我们对蛋白质-小分子相互作用的理解的举措,如结构基因组学联盟、酶功能计划、阐明可药用基因组计划和开放靶点等项目,在未充分研究的蛋白质的表征研究中正取得根本性进展。
在这种情况下,“功能表征”通常是指揭示与药物开发特别相关的蛋白质的分子特性,例如其结构、配体、被化学探针的可抑制性以及与疾病的关联。重点是药理学上方便研究的蛋白质家族,例如离子通道、G蛋白偶联受体和激酶等。但随着新方法(如PROTACs)的发展,可药用蛋白质的定义正在随着时间的推移而发展。
4、怎么用功能蛋白质组学解决注释非均一性问题?
需要区别两种不同类型的蛋白质注释工作:原创研究和“关联”法。对新型生物学功能的原创研究是必不可少的,其涉及许多详细的机制研究,耗时且花费巨大。对于研究人员来说,要致力于这样的工作,所研究的蛋白质必须具有一定的基础注释水平,否则,其功能假设就缺乏基础。而“功能关联”的注释可以通过知识转移提供所缺乏的基础,从而将以前未表征的蛋白质与经过充分研究的蛋白质及其生物学功能联系起来。
、蛋白质组学方法特别适合于在大规模范围内揭示功能关联。这包括鉴定蛋白质-蛋白质相互作用的技术,识别哪些蛋白质共同调控的方法以及揭示哪些蛋白质共享亚细胞空间的方法(具体见图2)。例如,在基于抗体的蛋白质组学鉴定出数百种额外的中心体蛋白之前,人们认为大多数中心体蛋白已被鉴定出来。
虽然基于质谱的蛋白质组学尚未达到基因组方法的基因覆盖率,但在研究(蛋白质编码)基因的功能时,直接观察蛋白质可能尤其有用。例如,比起mRNA共表达,蛋白质共表达在捕获功能关系时的作用尤其显著。基于蛋白质的分析还具有区分蛋白质形式的潜力,即被表达蛋白质的单个分子形式,其中剪接和翻译后修饰等显著增加了蛋白质组的功能多样性。蛋白质形式表征可能需要使用自上而下或自中而下的蛋白质组学方法。蛋白质组学的通量正在迅速增加,已经有方法可以每天在单台质谱仪上记录数百个蛋白质组。
然而,蛋白质注释非均一性不太可能完全通过大规模方法解决。努力共同解决蛋白质注释偏差的第一步可能是系统地为进行靶向相关实验的个体研究人员提供必要的最小数据基础,比如使用质谱法大规模鉴定蛋白质-蛋白质和蛋白质-蛋白质复合物相互作用的BioPlex和项目;使用抗体将人类蛋白质归属至不同的组织和亚细胞位置的人类蛋白质图谱(Human Protein Atlas);以及旨在通过蛋白质组学表征50种未充分研究蛋白质的neXt-CP50项目等。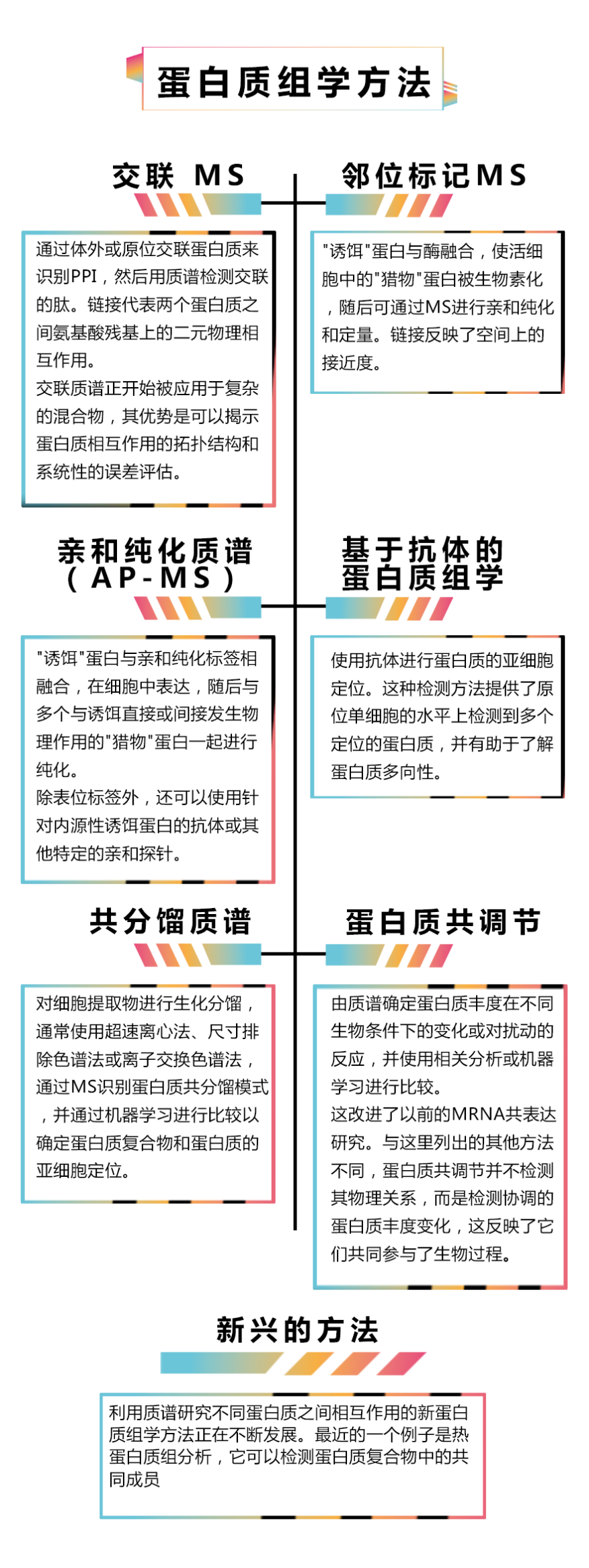 图2. 揭示不同蛋白质之间关联的蛋白质组学方法:质谱(MS)和基于抗体的方法,通过识别不同蛋白质之间的相互作用(PPI)来实现注释转移,在它们链接性质、可扩展性和偏差上有所不同,每种方法都有优势和劣势。以上是近年来的关键技术详单。值得注意的是,有各种非质谱的方法也能揭示不同蛋白质之间的关联。包括二元分析法,如Y2H、LUMIER、遗传相互作用筛选和代谢特征分析等。
图2. 揭示不同蛋白质之间关联的蛋白质组学方法:质谱(MS)和基于抗体的方法,通过识别不同蛋白质之间的相互作用(PPI)来实现注释转移,在它们链接性质、可扩展性和偏差上有所不同,每种方法都有优势和劣势。以上是近年来的关键技术详单。值得注意的是,有各种非质谱的方法也能揭示不同蛋白质之间的关联。包括二元分析法,如Y2H、LUMIER、遗传相互作用筛选和代谢特征分析等。
5、如何增加功能蛋白质组学在机制研究中的作用?
生物学家可以通过各种强有力且便捷的资源来查看分子网络,包括IntAct、BioGRID、NDEx和STRING。尽管这些资源很容易获取,但注释偏差却在不断加剧,原因可能很多:也许细胞生物学家不了解这些注释门户网站,也可能是缺乏信任,或者注释不足和对不同注释的整合不足等。
细胞生物学家可能会对依赖大规模项目的数据仍存疑虑,事实上,以统计学方式处理误差是大规模方法的独特优势。虽然误差无法避免,但其严重程度是了解结果可靠性的关键参数。功能性蛋白质组学技术中已经确定计算错误发现率(FDR)的一个例子是交联质谱法。同样,所有质谱法蛋白质鉴定的常规计算方法也需要计算FDR。此外,空间蛋白质组学中的统计框架也正在开发,以期将蛋白质分配到亚细胞区室。
除了扩大可用的大规模数据量外,无疑还需要开发新的工具和技术,填补目前方法留下的系统性空白。新兴功能蛋白质组学技术有交联质谱法、共聚合蛋白质组学和研究动态亚细胞区室的方法。现在,预测蛋白质结构取得了巨大成功,可能进一步改善基于结构的功能预测,尤其是在预测的结构可以被交联质谱法等实验证实时。这些技术和其他细胞内技术特别值得关注,因为许多蛋白质需要折叠或辅助因子或翻译后修饰的协助才能正常运作。此外,研究单细胞的蛋白质组技术正在日益成熟,该技术可以确定细胞间的异质性。
最后一个关键挑战是跨维度(时间和空间)整合不同类型的数据,这将最大限度地发挥不同类型组学数据之间的协同作用。比如人类蛋白质图谱和BioPlex数据的整合,表明了细胞层级结构的生成可以揭示许多新的细胞系统,而这些系统无法通过单独任何一个数据集检测到。这样的计算工具也可以通过提供数据驱动的假设来加速科学发展,也就是说,研究人员有望可以将他们的数据与蛋白质组学大数据联系起来。即使一个蛋白质的功能得到了很好的注释,也有越来越多的证据表明,一些蛋白质有能力执行其他不相关的功能,在文献中被称为“兼职”。由于研究人员假设过“一个蛋白只有一个功能”,因此人们没有为大多数蛋白寻找其它功能。对功能蛋白质组进行全系统梳理的另一个好处是,能提供充分研究的蛋白质的其它功能注释,并更好地了解蛋白质能够做哪些“兼职”。
6、如何量化功能表征的进度?
如果要制定出解决蛋白质注释非均一性的策略,并对其进行优化和评估,就需要保证信息可靠且足够。评估功能特征绝非易事,很大程度上是因为这个术语本身就有不同的含义。“蛋白质功能”可以指蛋白质广义上的生物学目的,比如它与哪种表型相关,或者它属于哪种代谢途径,也可以指如何理解蛋白质在分子水平中实现这些功能的结构和机制,例如酶的机制等。
确定蛋白质注释水平的方法已有很多,包括基于文本挖掘的文献分数, Uniprot注解得分,GO覆盖率的评估,以及根据蛋白质作为药物靶点的开发情况对其进行分类的系统。每一个指标都捕捉到或强调了现有注释中略微不同的方面,并不区分原始的表征和功能关联。
然而,为了系统地评估一个注释转移系统,有必要对其进行充分的量化研究以避免麦纳马拉谬误(根据一个单一的、容易测量的目标变量而不考虑更大范围和更难测量的因素来评估一个复杂目标的进展)。
7、如何避免以偏见换偏见?
我们认为,蛋白质组是注释基因功能的有力工具,但蛋白质组学方法也容易受到生物化学偏见的影响,例如来自蛋白质丰度和溶解度的影响。因此,为了系统地减少全基因组注释的偏差,可能需要单独优化多个功能蛋白质组学方法,并将其结果整合到一起。我们还可以将蛋白质组学数据与其他组学数据进行整合。例如,代谢组学可以捕捉到一个互补的功能谱。请注意,将蛋白质组学与遗传学、功能遗传学或代谢组学结合起来,可以大大提升表型预测效率。
然而,无论采取何种方法,必须消除标准实验室的限制性因素。最近的多生物体蛋白质组学调查表明,通过比较蛋白质组学,可能会有更多的蛋白质会得到表征,因为大量蛋白质功能在进化过程中得以保留,以及保留的蛋白质在不同生物体中有不同表现。许多组学技术结合基因组编辑可以直接应用于人类细胞的研究,这引发了人们担忧非人类生物体研究经费可能下降。尽管详细的统计数据表明,这些担忧目前也许并无根据。对广泛多样的生物进行研究,不仅让我们发现了青霉素、GFP和CRISPR/Cas9,而且还可能帮助我们捕捉了人类蛋白质组的功能谱。
8、“未充分研究蛋白质”计划
现在正是减少人类基因组和蛋白质组的注释非均一性的时候(图4)。“未充分研究的蛋白质计划”将包括各种不同的数据生成方法,开发整合框架,并通过适当的平台向研究人员提供注释。该项目不仅要解决技术问题,还要找到基因功能缺失的生物医学原因,如生长条件定义不准确、单一时间点研究,以及对于极少数遗传变异性低的实验室模型过度关注。蛋白质功能扩展研究也可能促进功能蛋白质组学的方法学发展,并可能将其延伸到其它物种。
第一步,必须明确定义目标。如果功能蛋白质组学的作用是推动对未充分表征蛋白质的机制研究,那么科学家们开始此类工作所需的最基本信息是什么?这个问题只能由在分子和机制方面详细阐明单个蛋白质在细胞中的功能的科学家才能回答。最终,实验室科学家和审核者的主观判断,决定了哪些蛋白质应该被详细研究。
为了解这些意见,我们最近发起了一项调查(),科研工作者可点击文末“阅读原文”,填写调查问卷。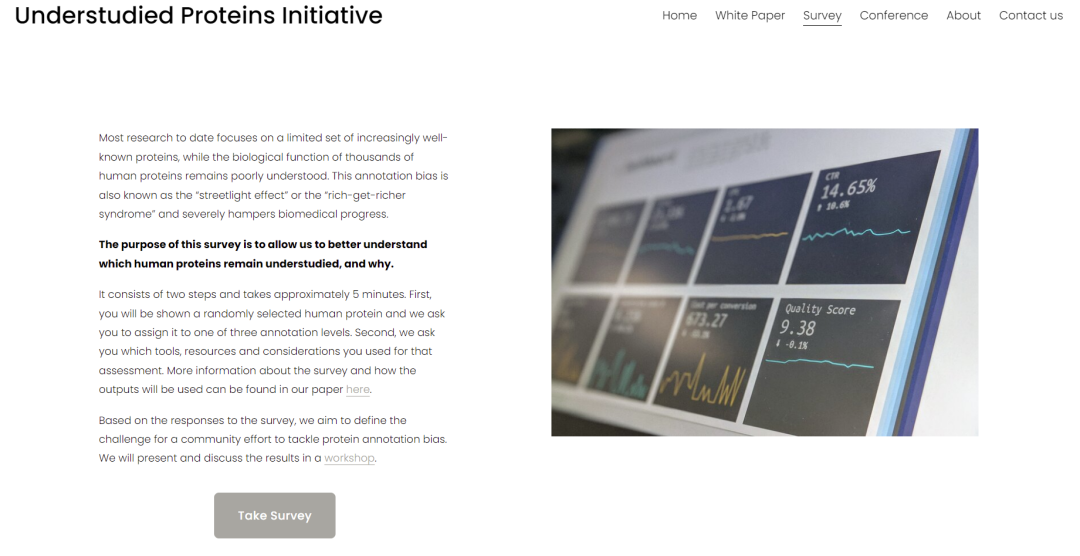 图3“未充分研究的蛋白质计划”调查截图 (欢迎各位科研工作者们点击文末“阅读原文”,进入页面后点击“Take Survey”参与调查)
图3“未充分研究的蛋白质计划”调查截图 (欢迎各位科研工作者们点击文末“阅读原文”,进入页面后点击“Take Survey”参与调查)
第二步,必须建立一个由有志于该领域的科学家组成的团队。该计划由惠康信托基金(Wellcome Trust)支持,并会在即将举行的会议()上启动。会议将讨论上述调查的结果,对“未充分研究蛋白质”计划目标的影响,以及如何监测这些目标的进展。这将助力开放式讨论,如,哪些技术或发展能够系统地释放目前生物医学研究中未表征的蛋白质的潜力?从而促进领域内更多发展。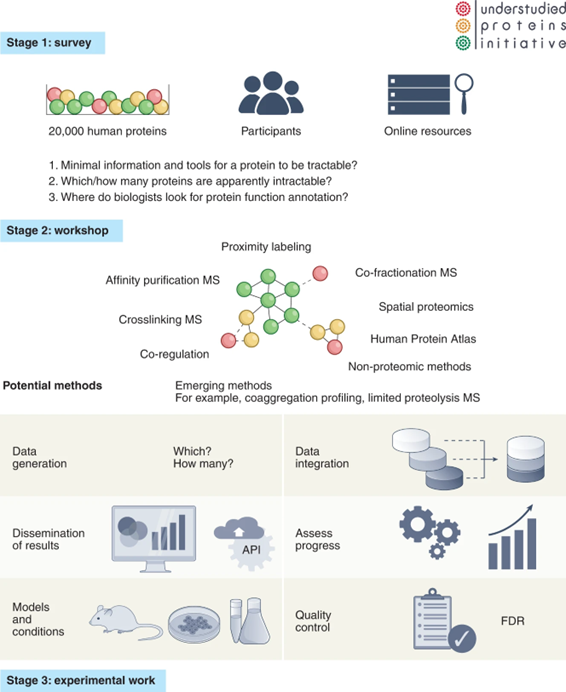
图4. “未充分研究蛋白质”计划的路线图。调查将有助于确定该计划的挑战和目标,研讨会将汇集来自大规模数据界的专家,建立涵盖六个行动领域的计划框架。最后,多个实验室将合作通过实验解决未充分研究蛋白质的问题。
附:“未充分研究的蛋白质计划”发起者及其科研单位:
- Georg Kustatscher: University of Edinburgh, Edinburgh, UK
- Tom Collins: Wellcome Trust, London, UK
- Anne-Claude Gingras: University of Toronto, Toronto, Ontario, Canada
- Tiannan Guo: Westlake University, Hangzhou, China
- Henning Hermjakob: EMBL-EBI, Cambridge, UK
- Trey Ideker: University of California San Diego, La Jolla, CA, USA
- Kathryn S. Lilley: University of Cambridge, Cambridge, UK
- Emma Lundberg: KTH-Royal Institute of Technology, Stockholm, Sweden
- Edward M. Marcotte: University of Texas at Austin, Austin, TX, USA
- Markus Ralser: Charité University Medicine, Berlin, Germany;
- Juri Rappsilber: Technische Universität Berlin, Berlin, Germany
参考文献
1. Kustatscher, G., Collins, T., Gingras, AC. et al. An open invitation to the Understudied Proteins Initiative. Nat Biotechnol (2022). Kustatscher, G., Collins, T., Gingras, AC. et al. Understudied proteins: opportunities and challenges for functional proteomics. Nat Methods (2022).

0
推荐
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}