“在我看来,《自然》杂志根本就不应该发表谷歌的这篇论文,因为它违反了FAIR(Findable可发现、Accessible可访问、Interoperable可互操作、Reusable可重用)的数据原则。……谷歌决定不共享用于生成模型的数据,甚至不共享模型结果本身。唯一共享的数据是模型最终识别出的稳定晶体,这让人难以复现模型。……我认为,像谷歌这样的公司参与科学进程固然重要,但也必须遵守同样的严谨标准。无论从哪个标准来看,一项无法被验证的工作都不能被视为科学。”
——Shyue Ping Ong(UCSD教授,Materials Project发起人)
撰文 | 刘淼、孟胜(中国科学院物理研究所/松山湖材料实验室)
巨头发力“AI+材料科学”
2023年11月底,Google旗下的DeepMind在Nature杂志发表了重磅论文,宣称他们开发了用于材料科学的人工智能强化学习模型Graph Networks for Materials Exploration (GNoME),并通过该模型和高通量第一性原理计算,寻找到了38万余个热力学稳定的晶体材料,相当于“为人类增加了800年的智力积累”,极大加快了发现新材料的研究速度(图1)。[1]

图1. Google旗下的DeepMind在Nature杂志发布了GNoME数据集及模型。
2023年12月,距离Google的GNoME模型发表数天后,微软发布了材料科学领域的人工智能生成模型MatterGen,可根据所需要的材料性质按需预测新材料结构。微软总裁在社交媒体上为自家大模型站台,评论道:“我们研发的MatterGen模型可以大幅提升新材料的按需研发效率”(图2)。[2]

图2. 微软总裁评论自家人工智能材料生成模型
2024年1月,微软与美国能源部下属的西北太平洋国家实验室(PNNL)合作,利用人工智能和高性能计算,从3200万种无机材料中筛选出了一种全固态电解质材料,完成了从预测到实验的闭环,该技术可助力下一代锂离子电池材料研发(图3)。[3]

图3. 微软的科学家从3200万种无机材料中筛选出全固态电解质材料,并实验验证。
材料科学无疑是一门重要科学,也是近代工业飞速发展的支柱学科。从石器时代到青铜时代,再到铁器时代,人类文明的各个演化阶段都和材料紧密相关。陶瓷为华夏文明的繁荣立下了汗马功劳;玻璃促成了光学器件的发明,为细胞生物学和天文学的进步打下了伏笔。可以说,人类文明发展史正是一部材料科学的演化史。
近期,人工智能技术的进步可谓是一日千里,卷到飞起。将人工智能方法引入科研已成为一个重要的交叉学科方向。除了Google和微软,Meta和字节跳动也在近期布局了相似的研发方向。Meta AI与美国高校合作,开发了行业顶级的催化材料数据集Open Catalyst Project和有机金属框架吸附数据集OpenDAC。一时间,科技巨头凭借自家的技术,将材料科学领域搅动到风起云涌。无机材料科学成为了他们的新赛道。
GNoME材料科学数据集详细解读
人工智能是如何变革材料研发的?科技巨头们看准了相同的技术路线:(1)通过理论计算获取材料科学数据;(2)通过高通量计算生产海量此类数据;(3)再将数据喂给人工智能模型;(4)借助模型推理未知材料的性能。这也意味着这是一种行之有效的技术方案,前景广阔。
人工智能是否将变革未来材料科学的研发方式?答案是肯定的。数据、算法、算力也将成为促成这场变革的核心因素。在铺天盖地的新闻和宣传中,让我们以Google发布的数据集为切入点,对其详细内容和逻辑一探究竟。
1. 继生物医药行业之后,材料科学是人工智能大举进入的下一个风口。数年前AI搅动生物和制药领域,美国的Schrödinger公司、Atomwise公司等众多企业的软件和模型让制药行业看到了新机会,在原子尺度筛选目标药物分子成为了各大药厂研发管线中的重要一环。
然而药物研发周期长,研发成本高,审批环节严格,因此已有部分AI制药公司转战材料科学。比如Schrödinger公司成立了材料科学部门。本质上,不论是生物医药还是物质科学,AI赋能背后的逻辑是一致的:通过人工智能方法,找到原子间相互作用的求解器和模拟器。
科技巨头们意识到,材料科学和制药有着相同的底层逻辑。万事具备,只欠“数据”。数据是人工智能起飞的助推剂,数据集的大小和质量高低直接决定了人工智能的预测本领。近期,得益于材料基因工程和若干材料科学数据库的发展,该领域已具备优质的数据资源,人工智能崛起的前提条件已经铺垫好了。
2. 数据集是人工智能大厦的地基。人工智能领域对数据的依赖度极高,数据集的覆盖度和质量直接决定了人工智能模型的高度。数据集的覆盖度决定了模型的泛化本领,数据集的一致性和可比较性决定了模型的预测精度。在人工智能数据、算法、算力三大要素中,数据是最具技术壁垒的环节。例如:GPT 3.5、Llama 2等大语言模型,即使开源模型源代码,也都选择不公布其数据集,没有优秀的数据集作为支撑,行业中的竞争对手很难训练出优秀的AI模型。
算法已经逐渐失去了技术壁垒作用,凭借算法引领行业一枝独秀的可能性微乎其微。
3. 理论计算为建立材料科学数据库立下了汗马功劳。密度泛函理论经过数十年的发展,已积累了成熟的技术储备,可以在短时间内生产出高度标准化的数据集。密度泛函理论通过求解体系中的电子运动方程,可以高效求解出化合物的性质,从而建立化合物中原子空间分布与化合物物性的联系。通过同时运行成百上千个计算作业,人们便可以生产出海量的数据集,目前材料科学领域使用最广泛的数据集,如Materials Project[4]、OQMD[5],都是基于密度泛函理论高通量计算获得的。GNoME数据集意味着Google已经掌握了材料科学的数据生产能力。
按照目前的材料科学研发技术,单凭实验数据积累,在数年内都无法企及类似的数据覆盖度和一致性。
4. Google的论文包含了GNoME模型代码和数据集两部分。数据集覆盖度和精度非常高。GNoME数据集从Materials Project衍生获得,采用了与Materials Project一致的计算标准和计算流程,因此可以和Materials Project[4]合并使用。Google称其通过高通量计算和密度泛函理论生产了220万种无机材料的计算数据,计算的同时通过主动学习不断预测热力学稳定的新材料,最终找到了38万种稳定的无机化合物,这无疑是对材料科学领域的巨大推动。
5. 虽然Google手握的GNoME数据集很大,涵盖220万种无机材料,但是随论文公布的信息仅包含很小部分的数据,即38万种无机化合物的结构、热力学稳定性及模型代码。Google至今未公开模型参数,因此第三方无法以开箱即用的方式运行模型的推理。Google也没有发布足量的数据,外界很难通过该数据集开展有效的模型训练。因此,Google是手握GNoME模型的独占方。
在未来的AI大模型建立过程中,数据是护城河,Google不开源完整数据,保证了其行业中不可被超越的领军地位。即使Google公开了38万个化合物的结构及热力学稳定性,但是google并没有公开很多关键信息,例如化合物的形成能(formation energy)。仅凭已公开的38万个材料数据,竞争对手也无法训练获得有效的模型。
数据生成环节是最费时费力的环节,但是目前行业里敢于迎难而上,通过各种方式生产标准化数据的机构、组织和研究者非常有限。大多数人期待搭乘“顺风车”,大家对数据共享满怀期待,却试图避开“数据生产难题”。
为了解决这个问题,行业中一种流行的观念是数据汇交,把各种孤岛数据“缝合”在一起,形成一个“大一统”的数据集。毫无疑问,这是一种寄希望于整合他人数据的方式,数年前就被推崇,但是目前尚未见到成功案例。例如,科技部的部分专项就有类似的数据汇交机制。
无疑科技巨头是清醒的,他们很清楚必须迎难而上,自己生产数据。他们大概率也没有意愿完整、大方地公开这些宝贵的数据集。这也是合情合理的,因为这些数据也许蕴含着巨大的商业价值。换个角度讲,开源、数据汇交的长期社会效益未必都是正向的。
6. 无机材料的相空间巨大,人类只发现了一小部分。本文作者详细分析了论文中38万种化合物的结构信息,发现其中30345种材料的元素组合(例:“Zr-Ti-Se”、“Ni-Te”)可以从Materials Project中找到,占比7.8%。这意味着在人类熟知的化学空间中,Google找到了30345个热力学稳定的材料。而大部分(92.2%)的稳定材料来自人类尚未涉足的元素组合(例如:“Rh-Ac”、“Zn-Cs”)。这意味着在未知的化学空间中,尚有很多未被发现的稳定化合物,人类已知的材料也许只是冰山一角。但是对于人类未涉足的化学空间,其中大部分化合物含有低丰度元素,此类材料的应用价值也是存疑的。(图4)
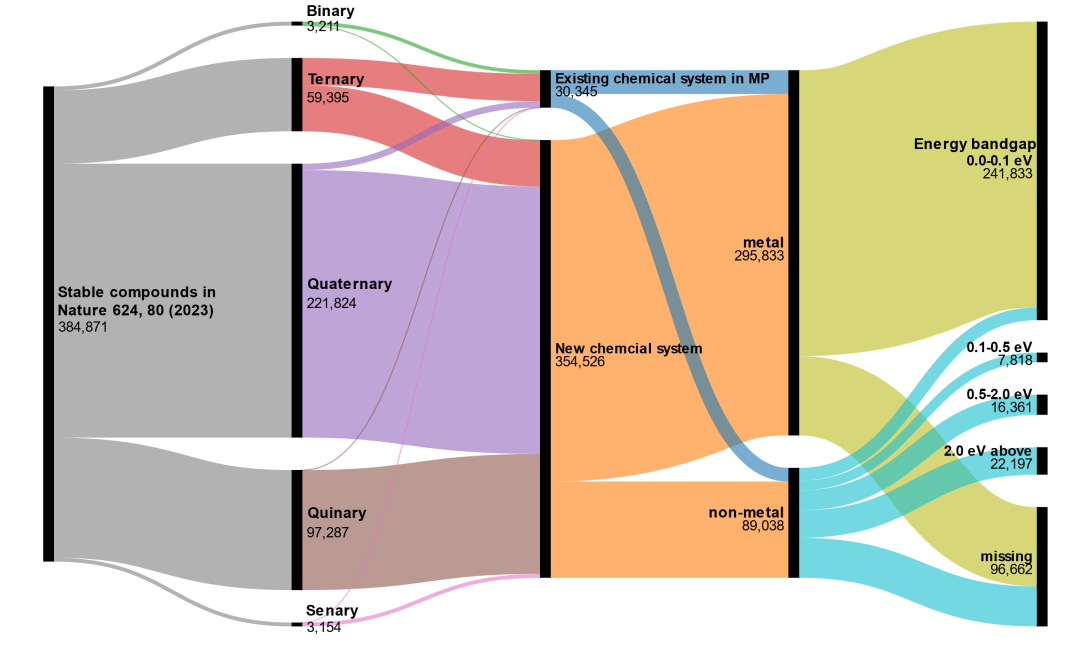
图4. Google的GNoME数据集详细分析。GNoME数据集宣称找到了384781种热力学稳定的无机材料。可以看到这些化合物中,三元、四元、五元化合物是主力。大部分化合物来自人类很少涉足的元素组合,且大部分是金属化合物。
7. GNoME模型在更广阔的化学空间中采样。该数据集覆盖了更广阔的结构空间和化学空间,因此是一个更加“广谱”的数据集,这对开发的AI模型非常有益。AI建模过程的本质是一种“求平均”,用行业术语表述,就是AI更善于求数据间的内插,而不是外推。
大家在衡量一个AI模型的好与坏时,通常的指标是预测精度,但是往往不谈模型的泛化本领。当然泛化本领的好坏也很难量化标定。提高泛化本领,需要更大、更广泛采样的数据集。
相较行业中通常以Materials Project数据为基础开发的AI模型(如CHGNET[6]、m3gnet[7]),GNoME模型拥有更“高一级”的数据集基础,必然具有得天独厚的泛化本领。
8. GNoME数据集是“严重偏科”的,金属材料占比60%以上。合金材料存在很多未知稳定结构是很正常的结果。因为金属原子之间容易结合形成金属键,进而降低体系能量,这是很常见的现象。然后这些金属元素在真实材料中大概率形成原子随机分布的合金相,而非GNoME数据集中的金属间化合物(intermetallic),因此大概率无法被合成。(图4&图5)
实际情况中,随便找几个金属元素混合,大概率都可以形成热力学稳定的合金,但这就算是发现新材料吗?如果算,从事合金研究的小伙伴们每天都在发现成千上万的新材料。
但对于人工智能模型训练,这些数据还是有重大意义的。
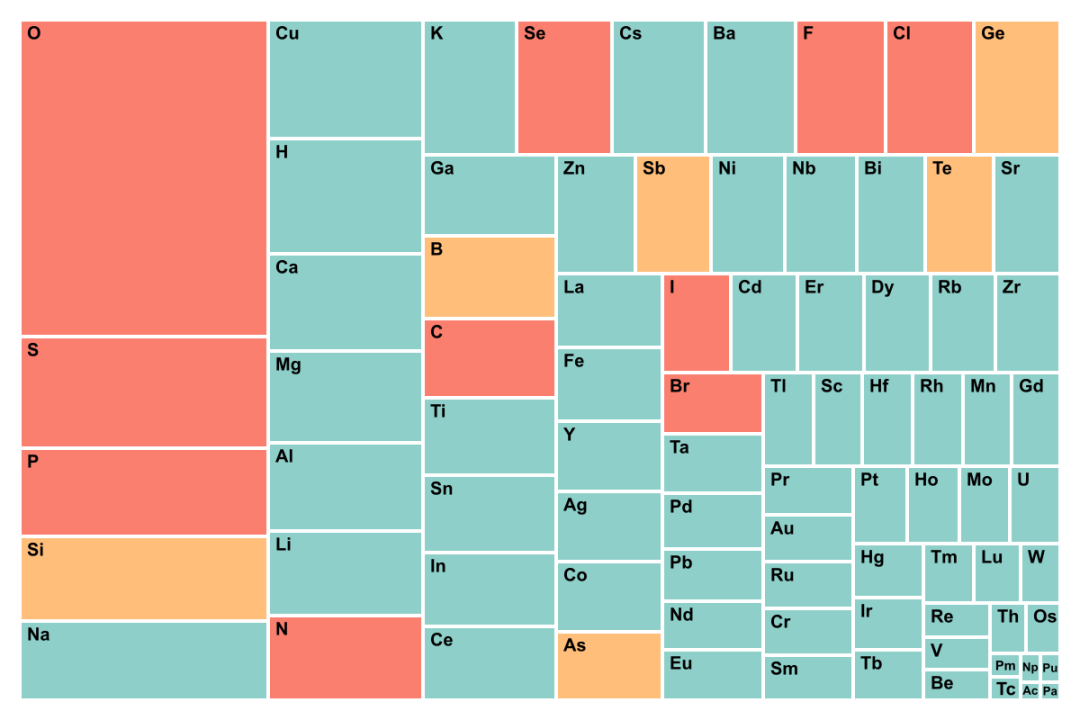
(a) GNoME
(b) Materials Project
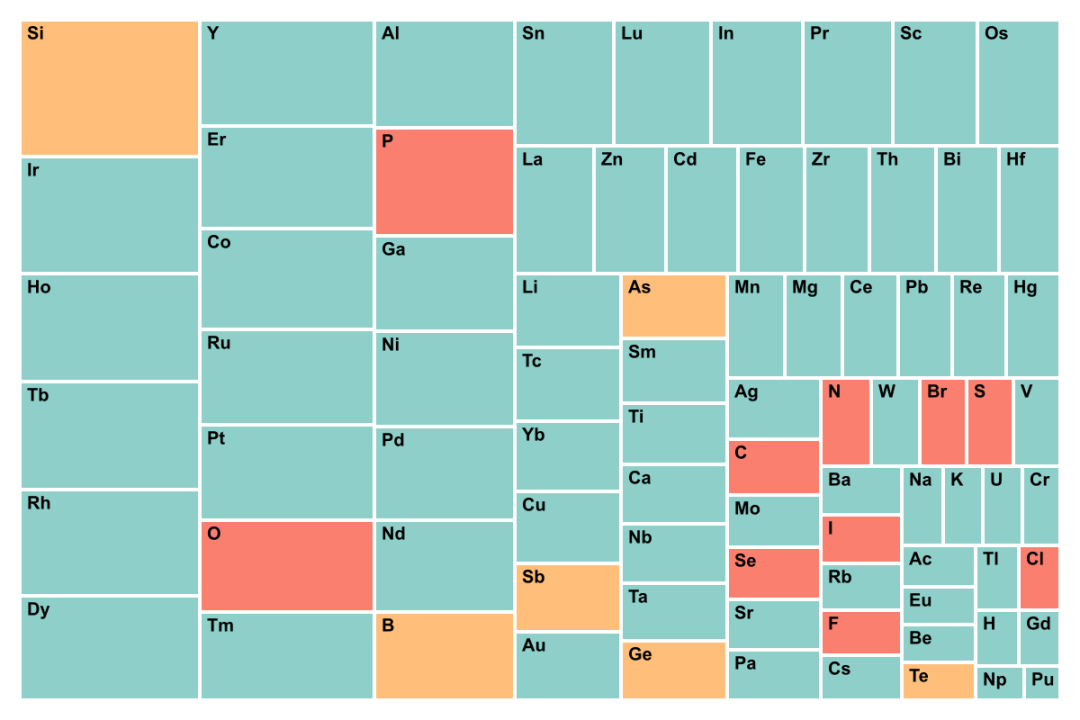
图5. GNoME数据集和Materials Project中元素出现概率的统计。GNoME数据主要探索低丰度元素,是人类较少涉足的化学空间。Materials Project探索的材料体系是较常见化学空间。
9. GNoME数据集中元素出现次数统计和Materials Project的统计结果非常不同。GNoME数据集中离子化合物的数量少,而金属元素,特别是低丰度元素出现的概率较大。如Ho、Tb、Rh、Er等出现的次数很多,而常见元素,如O、P、S出现的概率较小。这更加说明了,GNoME的采样空间是有偏颇的。(图5)
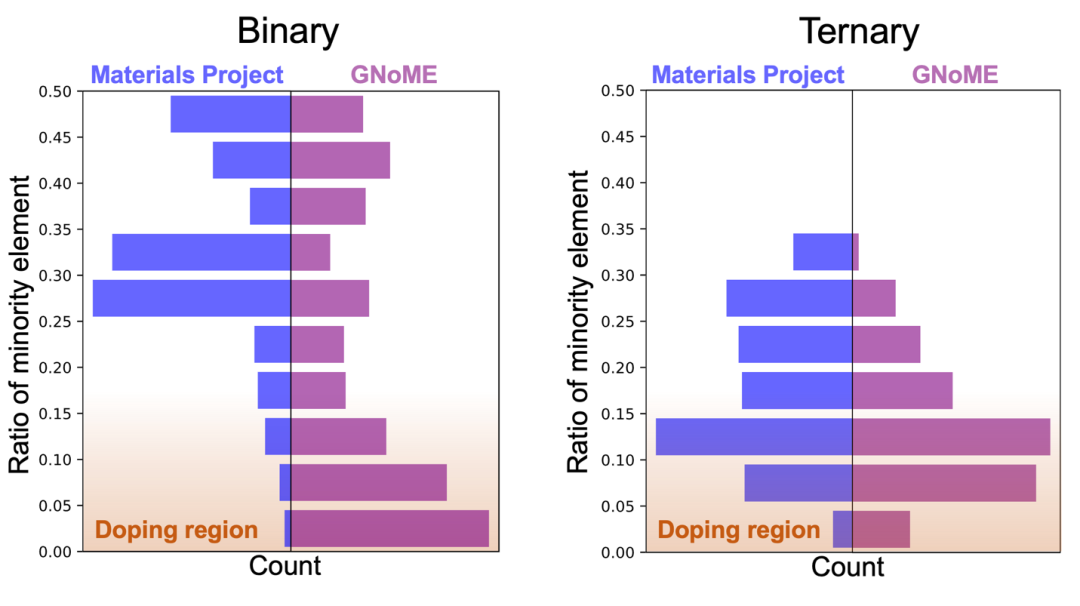
图6. GNoME数据集中,占比最低的元素的原子比例。比如MnO2中,Mn占比~33.3%,HBr35中H占比~2.8%。占比低代表化合物极有可能是一个掺杂化合物而非全新的稳定化合物。可以看到GNoME数据集中有很多疑似“掺杂化合物”,而非纯相,这一点在二元化合物中极为突出。
10. GNoME数据集中,除了多元金属间化合物占比大,掺杂结构的占比也很大,而此类结构也是比较难精确合成的。图6中可以看到化合物中占比最少元素的比例。可以看出一些热力学稳定的化合物是掺杂相。比如HBr35是在35个Br中掺一个H原子,但是实际中很难控制H在这个结构中周期性规则排布,形成与预测一致的HBr35晶体。该现象在二元化合物中突出一些,在三元、四元化合物中占比有所减少。(图6)
11. 所有视觉和语言模型中的先进算法,都将在物质科学中拥有用武之地。强化学习、注意力机制、扩散模型、预训练模型、多模态技术、生成算法、模型对齐机制、向量数据库等,迟早都将被不断引入材料科学中,并产生相应的工具。
未来任重道远,但充满希望
Google的GNoME数据集是“AI+材料科学”变革时代过程中的一粒火花。虽然数据集的许多具体细节仍没有发布,但无疑可以表明在尚未被人类涉足的化学空间中,仍有很多未知新材料等待人类去探索。数据集的发布在领域内开启了很多可能性。全球的研究人员将有机会进一步探索这些材料,可能应用这些数据创造出更多的人工智能应用、发现更多新材料。它不仅仅是一个数据集,它更是一张展示着可以重塑世界的无数创新的路线图。
在“AI+材料科学”的大潮中,数据是重中之重。生产数据集,特别是具有行业支撑作用的数据集,也许是一件“出力不讨好”的工作,但却是一场无法规避的“硬仗”。
注:本文精简形式的英文版已于2024年2月28日发表于Materials Futures。
DOI:10.1088/2752-5724/ad2e0c
URL:
参考文献
[1] A. Merchant, S. Batzner, S. S. Schoenholz, M. Aykol, G. Cheon, and E. D. Cubuk, “Scaling deep learning for materials discovery,” Nature, vol. 624, no. 7990, pp. 80–85, Dec. 2023, doi: 10.1038/s41586-023-06735-9.
[2] C. Zeni et al., “MatterGen: a generative model for inorganic materials design,” Dec. 2023, doi: 10.48550/arXiv.2312.03687.
[3] C. Chen et al., “Accelerating computational materials discovery with artificial intelligence and cloud high-performance computing: from large-scale screening to experimental validation,” Jan. 2024, [Online]. Available:
[4] A. Jain et al., “Commentary: The materials project: A materials genome approach to accelerating materials innovation,” APL Materials, vol. 1, no. 1. American Institute of Physics Inc., 2013. doi: 10.1063/1.4812323.
[5] J. E. Saal, S. Kirklin, M. Aykol, B. Meredig, and C. Wolverton, “Materials design and discovery with high-throughput density functional theory: The open quantum materials database (OQMD),” JOM, vol. 65, no. 11, pp. 1501–1509, Nov. 2013, doi: 10.1007/s11837-013-0755-4.
[6] B. Deng et al., “CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling,” Nat Mach Intell, vol. 5, no. 9, pp. 1031–1041, Sep. 2023, doi: 10.1038/s42256-023-00716-3.
[7] C. Chen and S. P. Ong, “A universal graph deep learning interatomic potential for the periodic table,” Nat Comput Sci, vol. 2, no. 11, pp. 718–728, Nov. 2022, doi: 10.1038/s43588-022-00349-3.
本文受科普中国·星空计划项目扶持
出品:中国科协科普部
监制:中国科学技术出版社有限公司、北京中科星河文化传媒有限公司

0
推荐
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}