近日,出现于南非的新冠病毒变异株Omicron(B.1.1.529)引发了全球前所未有的关注和担忧,多国对南部非洲国家实行航班禁入,有专家称Omicron是“最糟糕的变异株”,现有疫苗对Omicron 的效力还在观察之中。我们仍然面对着新冠的威胁。
除了推广疫苗的接种之外,积极推动各类抗病毒疗法的发展也是应对疫情持续扩散的重要举措。而当下可供感染者选择的治疗方案十分有限,迫切需要新型的特异性疗法,特别是口服药物,来预防更严重的感染、住院和死亡。同时,对患者及时有效的治疗也可以缩短传染期,降低大众患病风险。
撰文 | 汪汪
自新冠疫情暴发开始,人们就一直在曾经获批的药物中寻找有效的抗病毒药物,迄今为止,针对新冠病毒的小分子研究,已有超过500条药物研究数据以及400多项临床试验,其中绝大多数研究对象为已批准的药物,可惜老药新用的收效甚微,并没有出现令人满意的结果。虽然吉利德公司(Gilead)开发的瑞德西韦(Remdesivir)显示了一定的疗效,但是因为需要静脉给药,限制了其早期应用,在需要住院的病人中疗效有限 (详见《“人民的希望”尚未成真:瑞德西韦仍待验证》)。还有一些新型的口服药物已经取得积极的临床试验结果或者正在进行临床实验,比如作用于病毒 RNA 聚合酶的核苷类药物莫那匹韦和 AT-527[1]。
2021年11月5日,辉瑞公司(Pfizer)公布了其研发的新冠口服药物Paxlovid的三期临床研究结果,显示轻中度新冠患者在确诊三天内服用该药,其住院或死亡风险可降低约89%。Paxlovid的表现可以媲美新冠中和抗体的治疗效果,也显著优于前几日在英国获批上市的默沙东公司研发的广谱抗病毒药物莫那匹韦(Molnupiravir)。这一消息引起了人们的广泛关注,有媒体称Paxlovid为对付新冠感染的“银色子弹”(silver/magic bullet)。
新冠病毒的刺突蛋白仍在不断变异,令世界不得安宁。“银色子弹”能为人类打破这片可怖的阴影吗?
答案,可能藏在这款小分子抗病毒药物的研发历程中。
拨开迷雾,寻找可成药靶点
在新冠病毒出现以后,科学家们以非常快的速度解析了病毒的基因序列以及结构,为后续疫苗的设计和抗病毒药物的研发打下了坚实的基础。现在我们知道,新冠病毒是一种具有包膜的单正链RNA病毒,如图1所示,它由病毒RNA和四种结构蛋白组成:
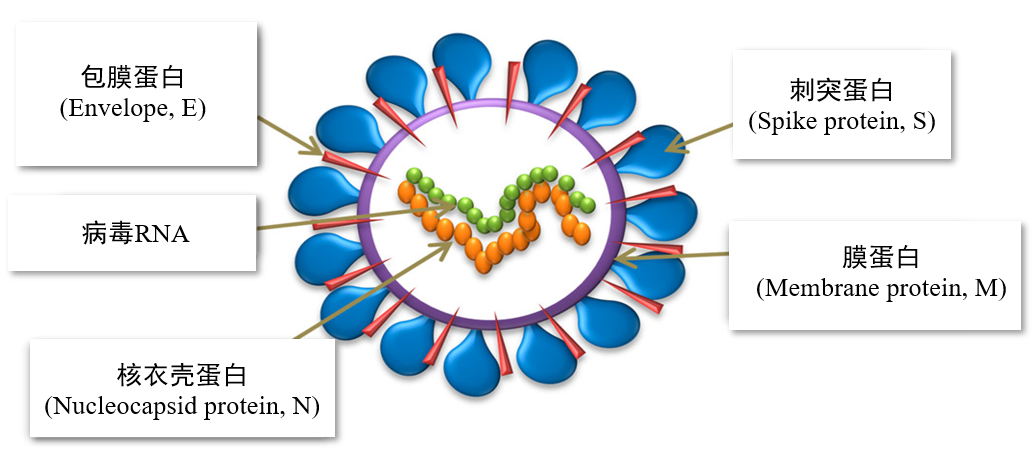
图1 SARS-CoV-2 及其结构蛋白示意图[2]
① 刺突蛋白(Spike protein, S)主要负责与宿主细胞受体结合,介导病毒的入侵;
② 包膜蛋白(Envelope, E)
③ 膜蛋白(Membrane protein, M),包膜蛋白和膜蛋白主要参与病毒复制装配和出芽过程;
④ 核衣壳蛋白(Nucleocapsid protein, N) 包裹病毒基因组,形成核蛋白复合体。
在病毒进入宿主细胞后,它会分解释放出病毒RNA,病毒RNA借由宿主细胞中的核糖体翻译成为两条多聚蛋白pp1a和pp1ab,这两条多聚蛋白就像是整盒包装的拼图,还不能发挥作用,需要在3CL蛋白酶和PL蛋白酶作用下拆装再重组,以形成具有功能的结构蛋白。用专业的话说,就是蛋白酶催化完成分子内水解,切割产生多个非结构蛋白,组成复制-转录复合物,以完成后续基因组的复制和结构蛋白的合成过程(图2)。
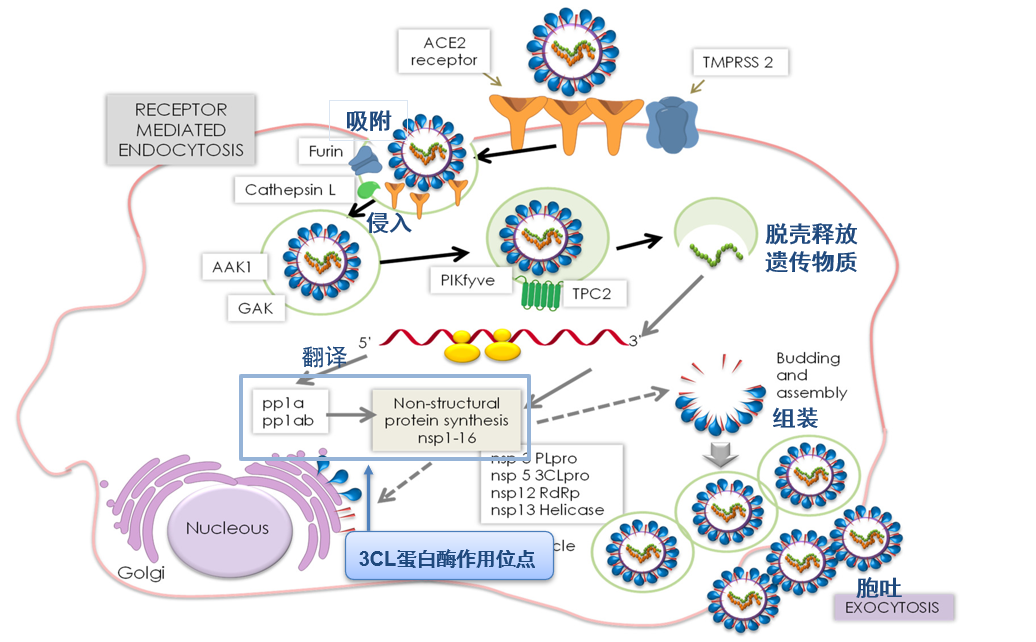
图2 SARS-CoV-2 感染宿主细胞循环示意图[2]
在这个过程中,3CL蛋白酶(3C-like protease)负责多聚蛋白11个位点的切割,以产生对病毒生存、繁殖非常重要的结构蛋白,因此又被称为主蛋白酶(Mpro)。我们可以设想,如果我们设计的小分子药物可以抑制3CL蛋白酶的活性,那我们就可以干扰病毒复制过程,达到抗病毒的效果[2]。科学家们当然也想到了这一点,因此当前对3CL蛋白酶抑制剂的研究非常热门。
过去,针对其他病毒进行小分子药物设计时,病毒蛋白酶也是非常重要的靶点。比如治疗人类免疫缺陷 (HIV) 和丙型肝炎 (HCV) 病毒的小分子口服疗法都曾以蛋白酶为靶点进行药物研发[3]。此外,最近的研究证明了口服3CL蛋白酶抑制剂在SARS-CoV-2感染的转基因小鼠模型中的药理活性[4]。这些都证明,3CL蛋白酶抑制剂具有非常大的潜力,可以成为抗病毒药物。
2020年4月,杨海涛、饶子和团队报道了新冠病毒 3CL蛋白酶与其抑制剂N3的晶体结构[5]。该蛋白酶为同源二聚体,每个单体由三个结构域组成(图3),在结构域Ⅰ和结构域Ⅱ之间形成底物结合口袋,底物结合口袋中,Cys-145和His-41形成催化二联体,能够结合并切割多聚蛋白。
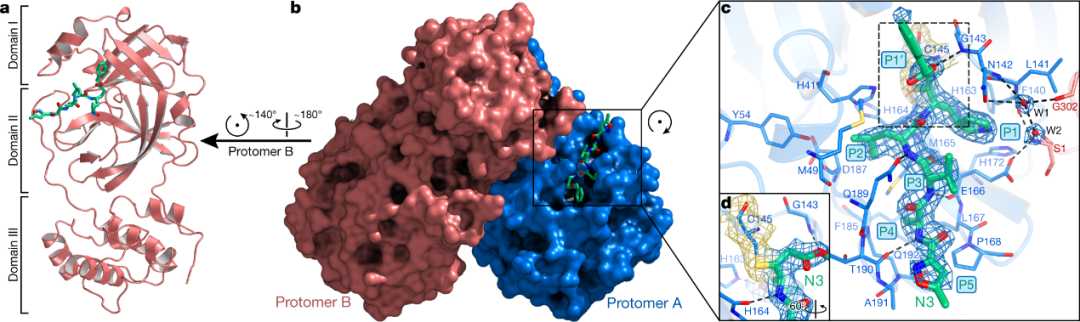
图3. SARS-CoV-2 3CL蛋白酶与抑制剂N3的晶体结构[5]
3CL蛋白酶在不同属冠状病毒间高度保守,在人体内没有同源蛋白。这意味着将其作为药物靶点进行抑制剂设计,不太可能“误伤”其他蛋白,可以达到较高的安全窗口或者治疗指数。
总的来说,由于3CL蛋白酶在病毒生存与繁殖过程中起着关键作用,而且与已知的人体内的蛋白酶没有显著的同源性,因此其抑制剂具有较好的选择性与安全性预期。并且,它不像疫苗或单克隆抗体药物那样,无法预防刺突蛋白的变体(新冠病毒的变异株层出不穷且具有极大威胁性),新冠病毒的3CL蛋白酶抑制剂已经成为非常有潜力、且具有代表性的小分子抗病毒药物设计思路,可用于设计抗病毒口服药物,治疗 COVID-19。
从SARS到SARS-CoV-2
2002年,SARS暴发。为应对疫情,辉瑞公司基于过去的工作研发出了化合物PF-00835231(图4)。在探索这个化合物抗SARS-CoV-1的机制时,科研人员采用基于荧光共振能量转移 (FRET) 的底物裂解实验,将其筛选确定为SARS-CoV-1 的3CL蛋白酶抑制剂。不过,SARS来得快去得快,随着2003年非典疫情结束,PF-00835231的研发计划就此搁浅[6]。
2020年初,新冠疫情在全球暴发,肆虐至今。大量的研究表明,引起新冠病毒病COVID-19的罪魁祸首SARS CoV-2与2002年引起SARS的SARS CoV-1具有密切的相关性——两种SARS冠状病毒基因组都编码两个重叠的多聚蛋白,在对冠状病毒复制至关重要的翻译后加工步骤中,它们在特定位点都被3CL蛋白酶切割。更为重要的是,而这两种病毒的3CL蛋白酶在与底物结合的催化域中的序列100%相同,这意味着对SARS-CoV-1 3CL蛋白酶有抑制作用的小分子对SARS-CoV-2 3CL蛋白酶应该也有抑制作用。
于是,辉瑞重新捡起化合物PF-00835231,推测其对SARS-CoV-2 的3CL蛋白酶也具有很好抑制效果。后续的研究结果证实了辉瑞科学家们的猜想,PF-00835231 表现出对SARS-CoV-2 3CL蛋白酶的强效抑制效果(抑制常数 (Ki) = 0.271 nM)。
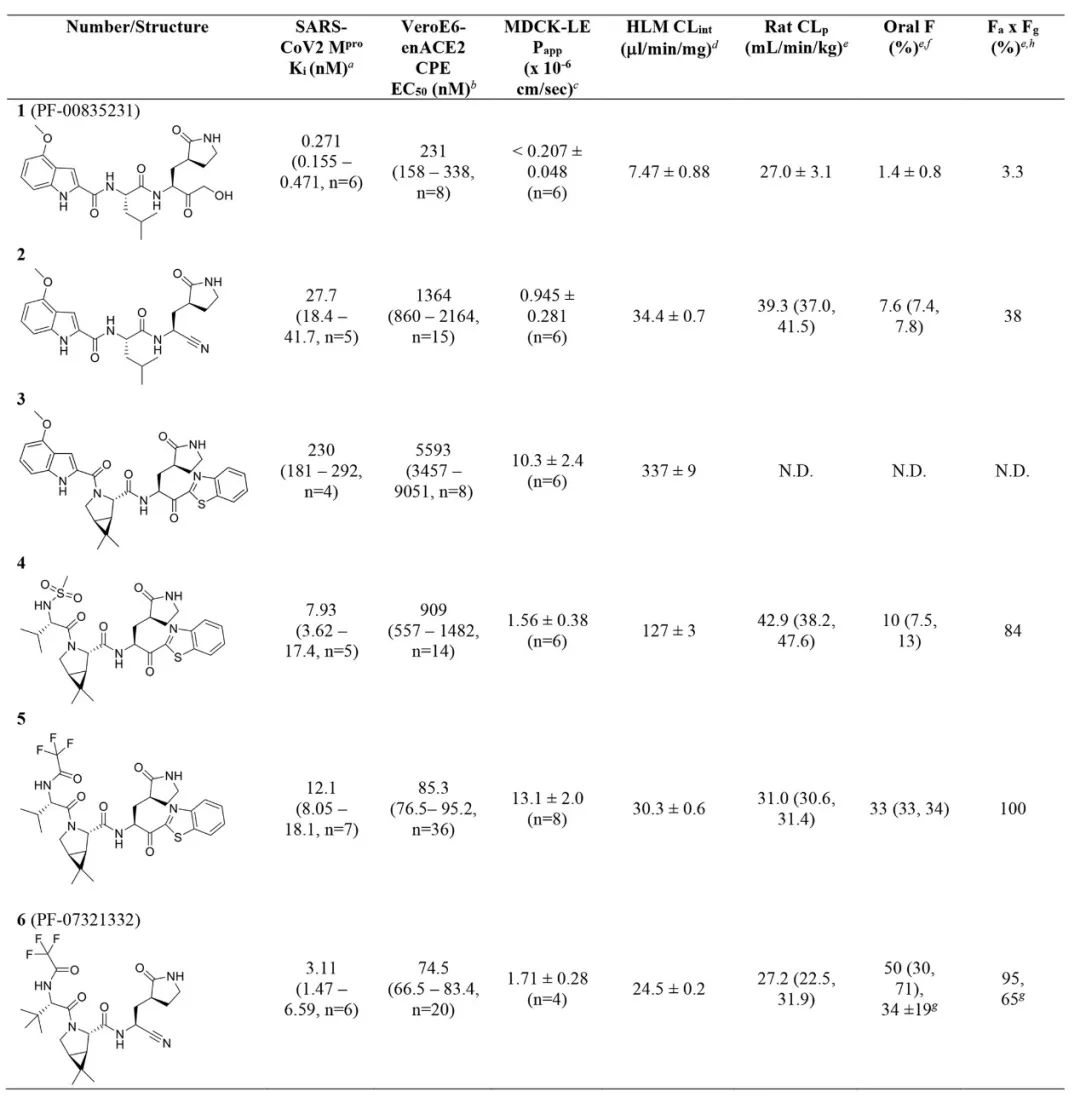
图4. SARS-CoV-2 3CL蛋白酶抑制剂的体外和体内参数。从左到右分别为3CL蛋白酶活性Ki、抗病毒活性EC50、通透性Papp、内在清除率CLint、血浆清除率CLp、口服生物利用度F和从胃肠道吸收的口服剂量的分数 (Fa x Fg)。[7]
如图4的数据第一行所示,PF-00835231具有优良的抗病毒活性和代谢稳定性,但其渗透性和口服利用度较低,因此很难进行口服给药,目前PF-00835231 的临床实验在采用它的磷酸盐前药形式 (PF-07304814) 作为 COVID-19 住院患者的静脉治疗选择[8]。
化合物改造
为了开发出适合口服的药物,辉瑞开始对化合物PF-00835231进行改造[7]。
一开始,我们先将PF-00835231称为“化合物1”。
科研人员首先考虑的是改善分子口服生物利用度,也就是提升药物进入体循环的程度。由于氢键供体较多往往会导致口服生物利用度较差,因此科研人员首先将化合物1的α-羟甲基酮改为氰基,得到化合物2;又在2的基础上同时引入氮杂双环和苯并噻唑,得到化合物3。
此时,相较于化合物1而言,2的口服利用度和胃肠道吸收的口服剂量分数都有了显著提高;化合物3的被动吸收渗透性也明显增强,但2,3的抗病毒活性相较于1又都有明显下降。
从分子与蛋白的结合模式分析可以看出(图5C),化合物3有效地填充了由 Met49、Met165 和His41 形成的亲脂性口袋,但不能与Gln189形成有效的氢键(如图5C所示),同时导致其活性远不如1。而且与化合物1相似,研究人员观察到这两个分子中的吲哚片段并没有很好地占据口袋。
因此研究人员进一步对3进行了结构改造,将吲哚片段改为甲基磺酰胺结构,得到了化合物4。相对于1和3,4可以与Gln189和Glu166都形成氢键,表现出更好的蛋白结合力、抗病毒活性和口服吸收效果。
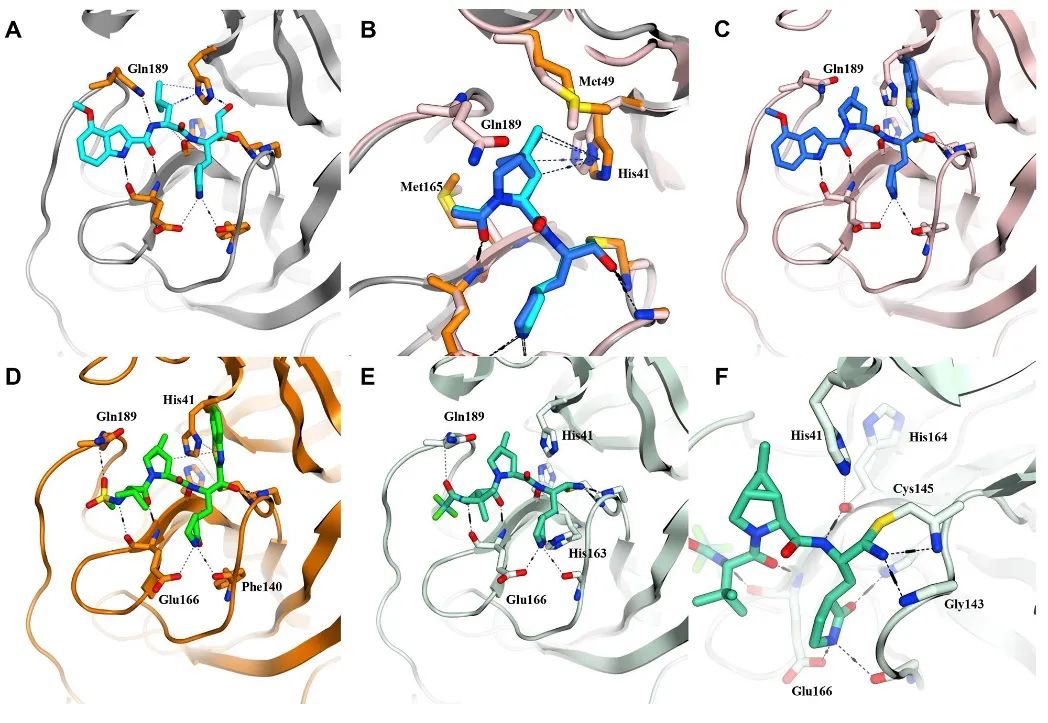
图5.(A)SARS-CoV-2-3CL蛋白酶与化合物1的共晶结构。(B)化合物3的双环模型与脯氨酸分子的构象叠加。(C)化合物3有效地填充了由 Met49、Met165 和His41 形成的亲脂性口袋,但不能与Gln189形成有效的氢键。(D)化合物4可以与Gln189和Glu166形成氢键。(E)临床候选 PF-07321332(化合物6)与 SARS-CoV-2-3CL蛋白酶结合的共晶结构。(F) Cys145与化合物6的氰基形成可逆共价结合。[7]
在化合物4的基础上,研究者进一步对结构进行优化,通过将甲基磺酰胺片段替换为三氟乙酰胺,得到了化合物5。化合物5和4对3CL蛋白酶抑制活性接近,但5在细胞模型中的抗病毒活性和渗透性都得到了显著的提升,同时化合物5在老鼠和猴模型的测试中,展现出了较好的药代动力学性质。
最后,对5的结构进行简化,保留其关键作用片段,将5的苯并噻唑基团替换为氰基,得到了化合物6,也就是此次改造所得到的临床分子PF-07321332(见图4最末行数据)。
化合物6相较于2-4,具有更好的抗病毒活性和更高的生物利用度。在化合物6与SARS-CoV-2-3CL蛋白酶的孵育实验中,研究人员发现,化合物6是可逆共价抑制剂,这意味着它比起一般的小分子共价抑制剂,脱靶带来的毒性风险更低了。综合考虑,化合物6具有结构相对简单、便于工业生产放大合成、溶解性更好、制剂难度更低等优点。因此。辉瑞最后选择了化合物6作为临床候选药物进行后续的开发。
除了可以抑制SARS-CoV-2-3CL蛋白酶的活性外,研究人员还发现化合物PF-07321332 具有广泛抑制冠状病毒3CL蛋白酶活性效果,包括 β 冠状病毒 (SARS-CoV-2、SARS-CoV-1、HKU1、OC43、MERS ) 以及 α-冠状病毒 (229E, NL63);它还表现出了对HIV-1 蛋白酶(一种病毒性天冬氨酰蛋白酶)的抑制活性。
在经过层层改造之后,科学家终于得到了具有更好抗病毒活性和更高生物利用度的化合物PF-07321332。综合考虑,PF-07321332具有结构相对简单、便于工业生产放大合成、溶解性更好、制剂难度更低等优点,是一个非常有前景的临床候选化合物。
药理学评价与基于药代性质的改造
在得到化合物6,也就是PF-07321332后,科研人员用两种与病理相关的细胞模型评估了PF-07321332的体外抗病毒活性:表达ACE2的人腺癌来源的肺泡基底上皮细胞(A549)和分化正常的人支气管上皮细胞(dNHBE)。两种细胞模型的结果都表明,PF-07321332对病毒复制有强烈抑制效果。
细胞水平评价之后,辉瑞进行了体内药效评价。
实验人员使用小鼠适应SARS-CoV-2的动物模型,评估PF-07321332的体内抗病毒活性,结果证明,在之前体外实验观察到的抗病毒浓度下,它在体内也可以有效降低感染鼠肺部病毒浓度,同时能够明显改善感染小鼠的多灶性肺部病变,这为化合物进一步向临床应用开发提供了有效依据。
另外,研究人员继续探究了化合物的临床前安全性和药物代谢特征。
实验结果表明,化合物PF-07321332具有较高的安全性和选择性,在体外的遗传毒性研究中,未显示出致突变或致裂变的毒性。在猴口服给药的模型中,化合物的生物利用度相对较低,这是因为,肝脏内的细胞色素 P450 3A4酶(简称CYP3A4)会氧化外源有机小分子,以便将其排出体外。这样一来,就降低了口服药物的利用度(很大部分被排出了)。另外,科研人员还观察到,PF-07321332的多个位点会被肝微粒体中的CYP3A4氧化代谢。这些研究表明,CYP3A4对PF-07321332代谢的影响至关重要,
因此,研究人员使用CYP3A4的抑制剂Ritonavir(RTV)作为共同给药,来改善PF-07321332的治疗效果。这个策略也是借鉴了以往的抗病毒药物给药策略,因为RTV是几个上市的蛋白酶抑制剂(如darunavir和lopinavir)的药物代谢动力学增强剂[9],它能使CYP3A4失活,减小主要抗病毒药物的代谢清除率,改善药物的生物利用度与药代动力学。
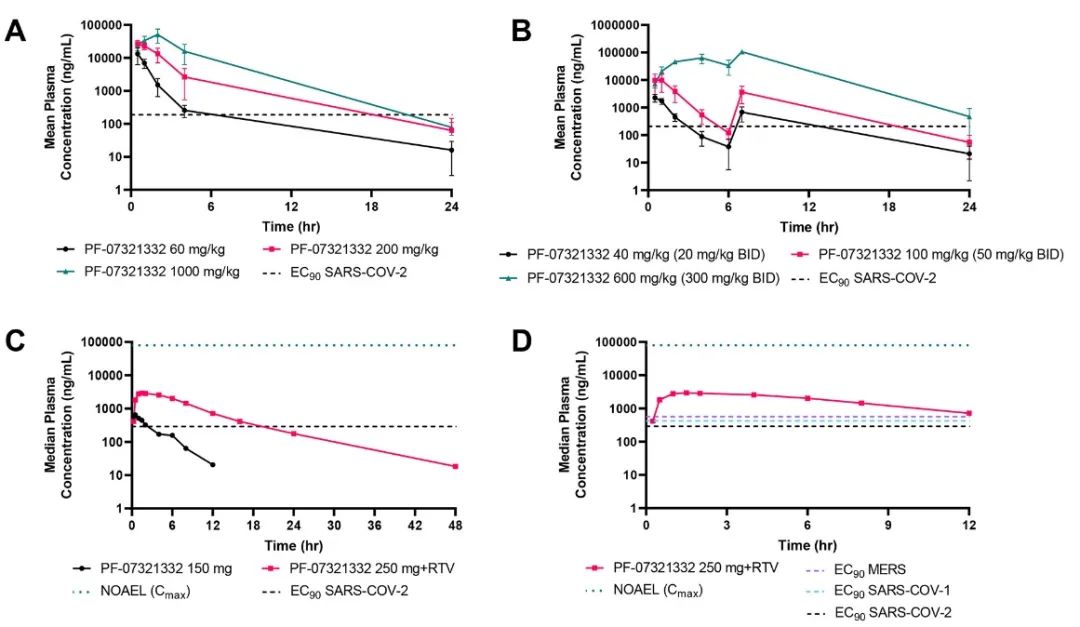
图6 PF-07321332的临床前毒理学和健康受试者单次递增剂量暴露研究。(A)每天给药一次PF-07321332的大鼠口服毒代动力学暴露(第14天)。(B)每天给药两次PF-07321332的猴子口服毒代动力学暴露(第15天)。(C)成人口服150mg PF-07321332和250mg PF-07321332+RTV的血浆浓度变化。(D)成人口服250mg PF-07321332和100mg RTV在12小时内的血浆浓度 。[7]
研究人员继续在健康受试者中,对PF-07321332单独给药和与RTV联合用药的安全性、耐受性和药代动力学进行了随机、双盲、安慰剂控制研究试验。在每一个测试剂量中,随机安排4位受试者接受积极治疗,2位接受安慰剂治疗。结果表明PF-07321332 具有较高的安全性及耐受性,当与 RTV 共同给药时,口服血浆浓度得到了显著增加且远高于SARS-CoV-2抗病毒的EC90 值(图6C,6D)。
种种结果表明,PF-07321332是一种可被口服吸收的SARS-CoV-2关键蛋白酶的抑制剂,具有非常好的体外抗病毒活性和优异的靶外选择性,以及较高的安全性,与 RTV 共同给药时,展示出了极佳的抗病毒效果和临床应用前景。因此,辉瑞推出了由Ritonavir和化合物PF-07321332组合而成的复方抗病毒药物Paxlovid,进行后续临床II/III 期研究。
11月5日,辉瑞公布了其新冠口服药物Paxlovid(化合物PF-07321332)的三期临床结果,数据表明,轻中度新冠患者在确诊三天内服用该药,住院或死亡风险可降低约89%。基于积极的临床结果,独立数据监查委员会建议提前结束临床试验,并向监管部门提交Paxlovid的上市申请。在结果公布的当天,辉瑞总裁Albert Bourla在一份声明中表示:“Paxlovid的出现将是我们抗击疫情中一个能够‘扭转局势’的事件(a game-changer)。”负责该药后期研发的一位辉瑞主管Annaliesa Anderson告诉《纽约时报》记者, “该结果比我们能够梦想到的最好的结果都好,希望该药帮助我们回归正常生活与终结疫情”。美国总统拜登也表示,该药有望成为我们手中用于对抗新冠的一个有效工具[10]。
辉瑞于11月16日正式向美国FDA提交了紧急情况下的上市申请[11]。
结语
Paxlovid的研发起始于辉瑞在SARS期间的工作,综合考虑结构、药代动力学性质、给药策略等多方面,经由研究人员巧妙的改造和完备的实验设计,最终得到了优良的临床效果。这是药物化学家、药理学家、与病毒学家多学科与研究组密切合作,遵循过往的经验与积累的知识,依靠计算机辅助,对先导分子进行成药性设计与改造,最后取得成功的一个经典案例。
新冠疫情暴发以来,科学家们马不停蹄,从前期的疫苗到现在的小分子药物,试图使用各种手段来遏制疫情的持续发展。辉瑞Paxlovid的出现,如同mRNA技术为代表的有效疫苗出现一样,很可能是人们抗疫过程中的一个标志性与分水岭事件。根据现有的数据,Paxlovid的出现,可以说是对付新冠病毒感染之特效药,堪比青霉素对抗细菌感染,有很大的希望让人们告别惧怕新冠感染的时代。因此,尽管疫情尚未结束,但我们依靠科学的力量在抗疫的道路上正取得一个又一个胜利与突破。通过控制传染源(如一定程度的隔离),阻断传播途径(口罩),保护易感人群(疫苗接种与有效的药物),我们应该能够控制疫情,回归正常的生活。
参考文献
[1] Good SS, Westover J, Jung KH, et al. AT-527, a Double Prodrug of a Guanosine Nucleotide Analog, Is a Potent Inhibitor of SARS-CoV-2 In Vitro and a Promising Oral Antiviral for Treatment of COVID-19. Antimicrob Agents Chemother. 2021;65(4):e02479-20. Published 2021 Mar 18. doi:10.1128/AAC.02479-20
[2] Gil C, Ginex T, Maestro I, et al. COVID-19: Drug Targets and Potential Treatments. J Med Chem. 2020;63(21):12359-12386. doi:10.1021/acs.jmedchem.0c00606
[3] Zhang L, Lin D, Kusov Y, et al. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J Med Chem. 2020;63(9):4562-4578. doi:10.1021/acs.jmedchem.9b01828
[4] Qiao J, Li YS, Zeng R, et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371(6536):1374-1378. doi:10.1126/science.abf1611
[5] Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289-293. doi:10.1038/s41586-020-2223-y
[6] Hoffman RL, Kania RS, Brothers MA, et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J Med Chem. 2020;63(21):12725-12747. doi:10.1021/acs.jmedchem.0c01063
[7] Owen, D. R., Allerton, C., Anderson, A. S., Aschenbrenner, L., Avery, M., Berritt, S., Boras, B., Cardin, R. D., Carlo, A., Coffman, K. J., Dantonio, A., Di, L., Eng, H., Ferre, R., Gajiwala, K. S., Gibson, S. A., Greasley, S. E., Hurst, B. L., Kadar, E. P., Kalgutkar, A. S., … Zhu, Y. (2021). An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science (New York, N.Y.), eabl4784. Advance online publication.
[8]
[9] Cvetkovic RS, Goa KL. Lopinavir/ritonavir: a review of its use in the management of HIV infection. Drugs. 2003;63(8):769-802. doi:10.2165/00003495-200363080-00004
[10]
[11]

0
推荐
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}