癌细胞与免疫系统之间的厮杀,还有太多谜团。每一次探寻,都带来新的希望。
撰文 | Samuel F. Bakhoum(纪念斯隆凯特琳癌症中心)
编译 | Kestrel
事情过去已有七年了,可当时失落的感受至今依然清晰。
那是2015年的一天,我拿到一位病人的肿瘤组织DNA测序结果,她的肺癌已经转移到了脑部,但是在报告里我找不到半点可以指明治疗靶标的基因改变,同时,另一件事引起了我的注意:数据显示,几乎每条染色体在结构上、数目上都发生了很多变化。
正常细胞中,每条染色体有两份拷贝,但癌细胞里面的染色体则不然,拷贝数目多少不定,少的只有一份,多的可以有五六份拷贝,有时候还会出现染色体碎片。这种基因组混乱的现象称为“染色体不稳定性”(chromosome instability),简称CIN。虽然癌症研究者和临床医生早就知道CIN是癌症进展到晚期或有转移的标志,但我从来没见过像这位病人这样,才59岁情况就这么严重的,更何况她才确诊不久,而一般认为CIN是在比较长的时间跨度上逐渐发生的。
肿瘤科医生对此束手无策,因为针对CIN没有太好的靶向疗法,病人只能简单地接受多轮全身化疗和放疗。我的病人也一样,除了一般性的副作用,她还得承受脑部放疗带来的神经毒性,可能会造成失忆和其它认知缺陷。
如此困境令我联想起自己的细胞生物学背景。过去,我读博期间的科研工作就是去搞清楚正常细胞有丝分裂过程中染色体是如何平均分配的。这个过程错综复杂,但每天都在许多组织中上演。生命体演化出多重备份机制,来确保这个过程不会出错。若万一出错,染色体数目异常的细胞也会被迅速清除。但癌细胞就不一样,它们对染色体异常的耐受性很强,且如此大尺度的基因变化与病程进展密切关联。然而,我们并不清楚,在肿瘤的发展和转移过程中,CIN是否扮演着积极的角色,又是怎样起作用的。
直到几年前,我开始着手研究CIN到底是会推动癌症发展,还是只是一种伴随癌症发生的现象。为此我与Lewis Cantley展开了合作,他当时是威尔·康奈尔医学院(Weill Cornell Medicine)迈耶癌症中心(Meyer Cancer Center)的主任,现供职于丹娜-法伯癌症研究所(Dana Farber Cancer Institute)。我们对多种染色体不稳定且具有转移性的癌细胞进行了遗传操控,降低它们的CIN水平,又不影响它们所携带的其它遗传学异常。结果非常明显,失去CIN的癌细胞同时也失去了转移的能力。还有一件事令我们惊讶,就是我们发现CIN是通过造成慢性炎症来促进癌细胞转移的。所以,归根结底,让癌细胞得以脱离原发性肿瘤而去入侵其它器官的,正是机体自身的免疫反应。
2018年,我们的工作发表在《自然》(Nature)杂志上。研究结果提示我们,让遗传物质变得不稳定这一步本身,对癌症的演进(evolve)至关重要。这可以成为新的治疗思路的出发点:我们能否在这一步中寻找靶点,来治疗CIN情况严重的癌症呢?能否想办法稳定基因组,或者减轻CIN造成的慢性炎症,来遏止癌症转移呢?能否改造免疫系统,让它去清除染色体数目异常的细胞呢?
针对这些关键问题,我在纪念斯隆凯特琳癌症中心(Memorial Sloan Kettering Cancer Center,MSK或MSKCC)的实验室采用了跨学科、基于细胞生物学同时结合单细胞基因组学、数学建模以及临床采样的方法。我们相信,通过整合这些方法,我们会理解CIN是怎样改变癌细胞的行为、提升癌细胞的适应性,来维持癌症发展的。进一步地,我们志在揭示是哪些细胞通路让癌细胞得以耐受CIN,然后通过靶向那些通路来治疗癌症。
也是在2018年,我和Cantley以及另一位同事Olivier Elemento联合创立了Volastra Therapeutics公司,以期相互补足我们在CIN方面的学术工作。公司的科研人员正在为多种癌症开发靶向CIN的疗法。通过这样广泛的合作,我们希望为染色体不稳定性癌症患者探索和开发新的治疗方法。
被忽视的癌症标志
自从2006年科学家测得首个癌细胞基因组以来,我们一直在不断深入了解是哪些遗传物质的改变在促使癌症发生、发展。科学家们开发出了一个个靶向疗法,作用于那些促进肿瘤发展的基因。这些疗法的理念都基于这样一个假设:如果我们能抑制这些基因,肿瘤就会停止恶化。因此,我们要对肿瘤组织进行基因组测序,找出每个病患身上的促癌基因,这一步已经成为MSK许多肿瘤科医生的常规做法。但是,当测序未能帮我们找到靶点时,肿瘤个体化治疗的局限性就显现出来了:的确有一些成功的病例,但是对大多数癌症晚期病人来说,效果还是十分有限。
即使靶向疗法可以派上用场,可能也只有一开始有效,因为肿瘤会“进化”。它们经常能逃逸我们的用药。而癌细胞最强大的武器之一,就是CIN。它们每一次分裂——托CIN的福——染色体都会发生随机重排。于是,染色体分离过程中发生的错误积累起来,导致癌变的组织里面各细胞在染色体组成和拷贝数上高度异质。这种现象称为“非整倍性”(aneuploidy)。的确,多轮治疗之后还是复发的晚期肿瘤,其特征就是染色体高度不稳定及非整倍性,对这样的肿瘤,抑制单个突变基因的药物已经起不了效果,尽管这些药物曾使癌症好转。
早在数十年前,研究者就已知道非整倍性是人类癌症的一个特征,但直到1997年,约翰·霍普金斯大学医学院(Johns Hopkins University School of Medicine)的Christoph Lengauer和Bert Volgelstein才首次证明CIN在促进癌细胞异质性方面的作用。通过他们的工作,人们很快就理解到CIN有刺激肿瘤演进、恶化的潜力:CIN能够调节染色体拷贝数,因而也调节这些染色体上基因的拷贝数。近期,哈佛大学医学院的Stephen Elledge发现,人类的恶性肿瘤的确会尽可能地增加带有致癌基因的染色体拷贝数,并减少带有抑癌基因的染色体的拷贝数,从而提高自身的适应性。
尽管CIN对人类癌症来说很重要,但实验室还是聚焦在基因突变上。新一代测序技术所带来的方法学革命引导学界把目光都放在了单个基因对癌症发生的贡献上,并由此得到了许多重要的发现,扩展了我们的认识,使我们了解了许多基因在肿瘤发生过程中所起的作用。但是,这种方法也有弊端,它忽视了大尺度染色体畸变及其对基因功能和癌细胞行为的影响。从整块肿瘤组织样本中纯化出DNA,对其进行测序,固然能够让我们更清楚地看到染色体上的遗传信息,但无法将序列发生改变的DNA片段定位到染色体上,而且模糊了细胞间染色体拷贝数的异质性。
在过去的十年间,研究者开始将更多注意力放到大尺度的染色体改变上。2010年,MSK的Robert Benezra等人发表了一项重要研究,表明已经获得CIN的肿瘤不再依赖一开始引发癌症的致癌基因。具体来说,当研究者上调致癌基因KRAS诱导小鼠形成肺癌后,再取消上调,能观察到肿瘤消褪;但如果采用基因工程手段,进一步往癌变的细胞中人工引入CIN,肿瘤消褪现象就观察不到了。所谓靶向治疗,就是去特异性地抑制像KRAS这样的致癌基因,不让它们驱动肿瘤发生,但恶性肿瘤会逐渐对这种疗法产生抗性,这项工作就告诉了我们抗性是如何产生的。
此后,伦敦大学学院(University College London)及克里克研究所(Crick Institute)的Charles Swanton组给出了强有力的证据,表明CIN在人类癌症中十分重要。2017年,通过随访肺癌病人,Swanton团队证明了是染色体不稳定——而不是肿瘤基因组中点突变的个数——与整体生存率的降低相关。他们后续的研究显示,无论是肿瘤转移还是癌细胞逃逸免疫监控,CIN很可能在肿瘤生物学的方方面面都扮演着重要的角色,癌细胞每次分裂伴随的染色体拷贝数逐步改变,为肿瘤提供了能在各种选择压力下演进的能力。
正因为这些科学家们不断研究CIN在癌症中的作用,癌症基因组学与细胞生物学之间的界限逐渐在消解。一方面,染色体所携带的遗传密码可以用复杂的基因组学手段破译;另一方面,染色体生命周期与细胞分裂过程中的分离行为基本上可以用分辨率较高的光学显微镜来跟踪。例如,分离过程中出现错误的染色体最终会出现在一个容纳小段DNA的“微核”(micro-nuclei)中,与细胞核——即“主核”——分离。微核一直被看作是区分癌细胞及周围正常组织的一个特征。多个研究组的工作显示,包裹微核的核膜经常破裂,把染色体洒到细胞质中,暴露在核酸酶下,同其它蛋白质一样被降解,变得四分五裂。
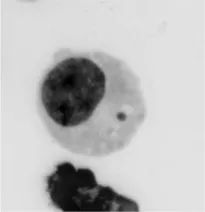
图1. 微核的显微照片。细胞中的小黑点即是微核。(https://doi.org/10.1016/B978-0-12-800764-8.00006-9)
发生大规模的染色体断裂之后,有些片段会丢失,有些则会随机接在一起,那么片段与片段之间连接的方向和顺序都可能出错,从而形成新的、高度异常的染色体。这个过程称为“染色体碎裂”(chromothripsis)。近期,研究者发现,染色体碎裂是刺激癌症恶化的重要机制。除了染色体数目一步步变化外,通过大规模的染色体重排,碎裂的染色体很快就形成一个能开出癌症的大奖池——癌症一发生,就已经很严重了。这个过程有可能快速扩增致癌基因,并丢失抑癌基因。我们现在还知道,染色体碎裂可能会把致癌基因置于高度活跃的染色体区域附近,还会促进环状染色体外DNA的形成,两者都会促使肿瘤细胞快速形成对靶向治疗的抗性。
尽管研究者很早就发现了癌症中染色体的混乱状态,但直到最先进的基因组学和显微技术出现,我们对染色体碎裂过程的理解才逐渐清晰起来。2015年,丹娜-法伯癌症研究所的David Pellman及同事应用显微技术捕捉到了出现染色体分离错误的单个癌细胞,并对其进行了基因组分析。通过这种方法(他们称为Look-seq),研究者展示了在人类癌症基因组中常见的复杂染色体重排模式是怎样在一个细胞周期内出现的。通过各种各样的途径,CIN似乎既可以促进渐进式的,也可以促进间断性爆发式的肿瘤基因组演化。这,大概就是我那位病人的情况——确诊没多久就出现了大规模的染色体畸变。
微核的形成机制
细胞分裂过程中,有许多染色体分离错误都可能导致微核形成。即使最终染色体并没有错误分离,也还是有可能形成微核。例如图2所示的情况,虽然染色体最终是平均分配给了两个子细胞,但由于分离滞后,左边的子细胞还是形成了微核。这些事件不互斥,也不独立,只是每一次发生都会加剧染色体的混乱。
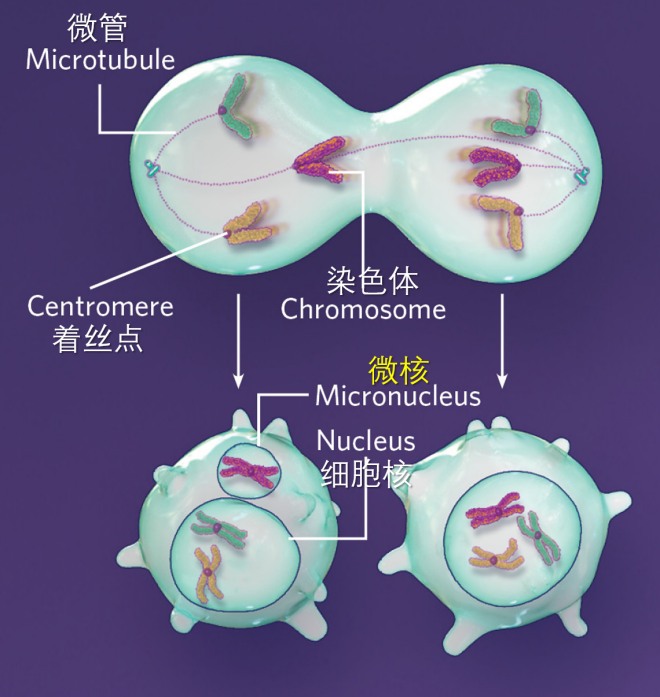
图2.错误附着。
当分裂细胞两极的微管附着到同一中心粒时(上),被附着染色体的分离会滞后于其它染色体,之后常常会被包裹到微核中,即使它最终还是分到了应该分去的细胞(左下)。
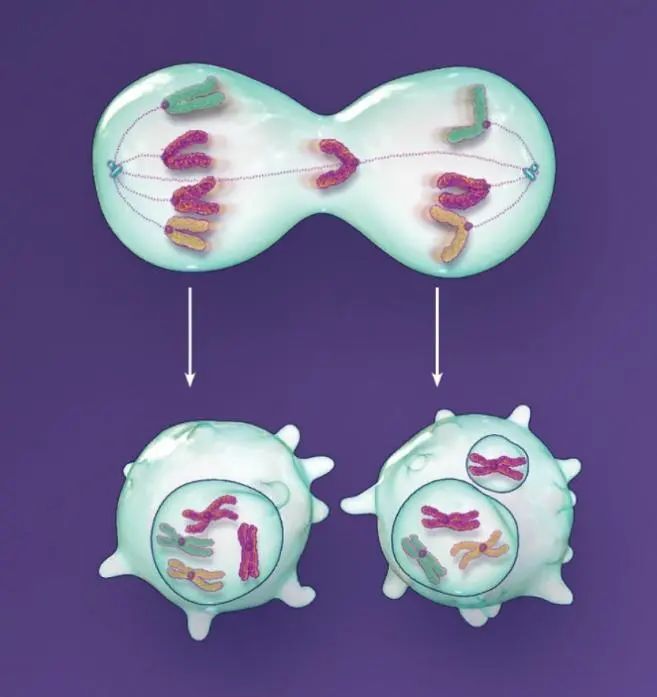
图2.非整倍性。
如果染色体分离真的出错了,不管是由于微管错误附着还是由于其它的原因,错误分离的染色体可能会随其它染色体被核膜包裹。要是没有被包裹,形成的非整倍体细胞就可能带有一条滞后的染色体,增加微核形成的风险。
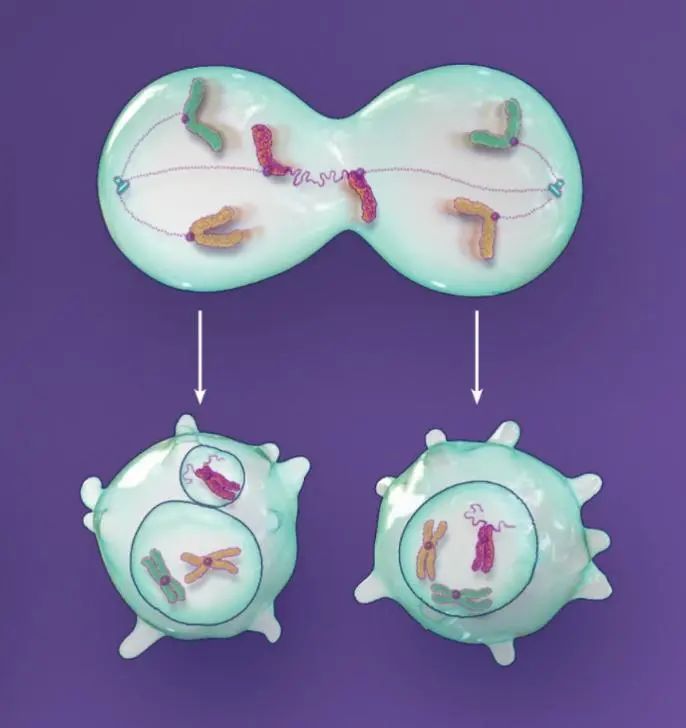
图3.染色体融合。
端粒的缩短或损坏能让染色体融合更有可能发生。这样的融合事件会产生带有两个中心粒的染色体。在下一次细胞分裂过程中,带双中心粒的染色体会被撕裂,分到两个子细胞中。这些损坏的染色体要么立即被隔离到微核中,要么在下一次细胞分裂过程中由于无法正常完成复制,还是被隔离到微核中。
CIN与炎症
在研究染色体不稳定性会怎样影响癌症转移时,我们得到了意料之外的发现。具体来说,已经发生CIN的癌细胞,它的炎症相关信号通路是激活的,这就能让癌细胞产生、分泌多种与癌症转移相关的炎症因子。这件事情一开始挺迷的,因为这些细胞只是在实验室里养着,并没有种到实验动物体内,也就没有任何接触到免疫细胞的机会。那么这些“炎症反应”是什么引发的呢?
在显微镜里苦寻良久,我们不仅观察到,发生CIN的细胞内,微核占了主体,还看到那些包含破裂微核的细胞带有一种与免疫相关的酶,叫做cGAS。cGAS于2013年首次被德克萨斯大学西南医学中心(University of Texas Southwestern)的James Chen(陈志坚)发现,它是一种定位于细胞质的双链DNA感受器。于是我们设想,微核破裂后,随之而来暴露在细胞质中的染色体可能会被癌细胞识别为危险信号,就像细胞识别出入侵的病原DNA那样。当然,随后我们便确认了,破裂的微核能够强有力地激活cGAS及其相关蛋白STING,进而激活固有免疫应答。但不像几天之内就能清除的急性病毒感染,癌细胞的细胞质中,微核破裂事件一起接着一起,导致炎症通路一直处于激活状态,炎症一直持续。
至此,事情已经很清楚了:癌细胞一定是利用了某一条保护性的免疫通路,才能够冲破这些炎症防线。虽然在肿瘤发生早期,固有免疫信号通路的激活可能保护人体,预防肿瘤出现,但到了一定阶段,肿瘤细胞能够越过这些保护性机制,耐受CIN所引发的炎症反应,并逐渐利用这些通路来驱动肿瘤生长。癌细胞维持炎症的能力对其向另一器官转移至关重要。免疫细胞是机体内最机动的细胞,人体遭遇感染或出现伤口后的数小时内,它们就能穿过血管系统迁移到静水压升高的发炎组织中,到达受损区域。癌细胞就是利用CIN和其它基因组异常,通过模拟这一生理过程来实现转移。
慢性炎症与癌症之间的关联由来已久。事实上,古罗马百科全书派学者Aulus Cornelis Celsus所描述的炎症最核心的特征对癌症也适用——发红、发热、疼痛肿胀,几百年来,临床医生常常将肿瘤称为“无法愈合的伤口”,因为它会持续不断地发炎。我们还不十分清楚炎症信号通路对癌症的发展起着什么样的作用,但是将内在的基因组异常(例如染色体不稳定)与癌症中持续的炎症关联起来,就能够发现,CIN不仅能驱动肿瘤的遗传异质性,也能通过非遗传性的机制(即模拟炎症反应)刺激癌症转移。
微核破裂如何促进癌症发展?
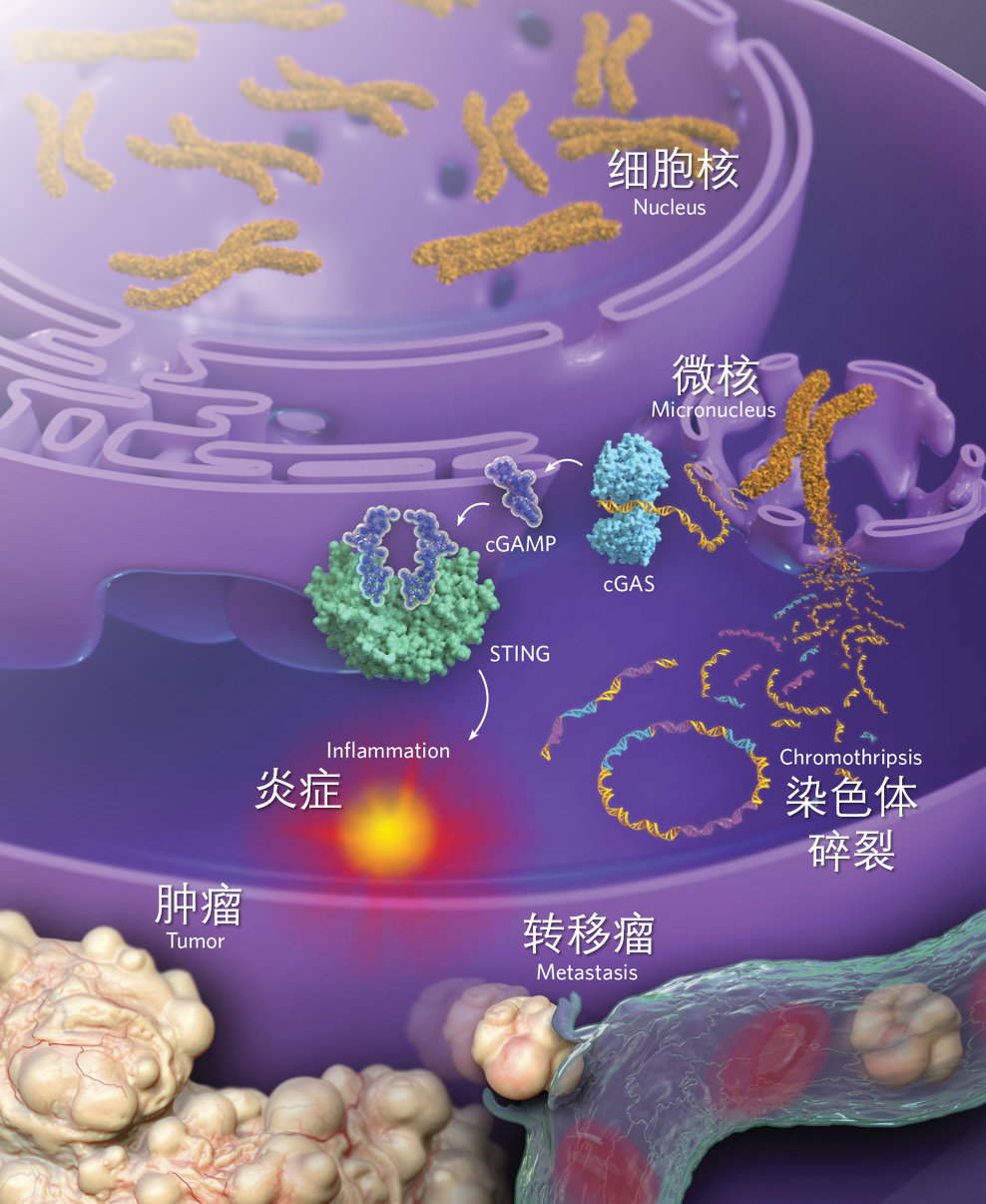
图4.微核破裂促进癌症发展。微核的核膜很脆,经常破裂,导致染色体散落到细胞质中。细胞质里面的染色体会被核酸酶切成小的片段,这些片段要么丢失,要么被随机连接到一起,要么首尾相连,形成环状染色体外DNA。这个过程就是所谓的“染色体碎裂”。这个过程形成的复杂的重排染色体会驱动肿瘤的产生。与此同时,停留在细胞质中的DNA会引发cGAS-STING炎症通路。一般认为,这条通路是从抵抗病毒感染的机制演化而来的。cGAS与微核破裂散落的DNA结合,催化生成2’3’-环鸟腺苷酸(cGAMP),从而激活STING蛋白及下游炎症通路。由于癌组织中有很多微核,就有可能一直激活这条通路,引发持续的炎症反应,驱动肿瘤生长和转移。
瞄准CIN
和癌细胞不同,正常的细胞无法耐受染色体分离错误。麻省理工学院(MIT)已故的Angelika Amon曾领导的研究发现,非整倍性与多重细胞缺陷有关联,例如代谢紊乱和线粒体功能紊乱,以及蛋白质折叠错误诱导的细胞应激反应。实际上,人体已经演化出多种机制,能够最终清除非整倍体细胞。达特茅斯盖泽尔医学院(Geisel School of Medicine at Dartmouth)的Duan Compton等人发现,应对染色体分离错误,正常细胞会快速激活肿瘤抑制因子p53,停止细胞分裂,阻止非整倍体细胞蔓延。这些重要的保护机制是为了维护基因组的完整性,一般有了它们,基因组就没事。但是在癌细胞中,这些防线被突破了。因此,理解肿瘤细胞是怎样应对CIN的,可能会对治疗癌症有启发。
学界对肿瘤细胞耐受CIN的机制表现出越来越多的兴趣。近期,多个课题组通过遗传筛选发现了一些基因和一些细胞活动,它们对CIN程度高的肿瘤细胞不可或缺,缺乏则会致死。其中一个是驱动蛋白Kif18a,在有丝分裂期间、染色体运动过程中起作用。对带有CIN的癌细胞来说,Kif18a蛋白在细胞分裂中必不可少,但是不带CIN的癌细胞就不然。有趣的是,如果一只小鼠缺乏有功能的Kif18a蛋白,它仍可以存活,只是表现出一些小的缺陷。那么这个驱动蛋白就有可能成为一个安全有效的治疗靶点。现在已经有一项一期临床试验在癌症晚期病人中测试Kif18a抑制剂的疗效。
还有多个课题组正在探索另一种治疗策略,即,抑制那些让肿瘤细胞克服慢性炎症的靶点。比如ENPP1蛋白,它一开始由哈佛医学院的Timothy Mitchison及斯坦福大学的Lingyin Li(当时也在哈佛)找到,后来我们组发现,它在染色体不稳定的癌细胞中会被选择性上调。ENPP1蛋白是定位在癌细胞外表面的酶,能降解会激发免疫反应的信号分子cGAMP。胞外的cGAMP被降解后,免疫细胞就不能发现癌细胞了。cGAMP降解还能产生腺苷,腺苷又加剧免疫紊乱,促进癌细胞迁移。癌细胞化敌为友、将免疫系统的保护机制挪作己用的能力让我们刮目相看。
Volastra公司的研究者们正在不断深入认识CIN的生物机制,再结合计算和遗传筛选的方法,慢慢发现一些癌症治疗策略。目前首要候选药物的靶标作用于微管结合染色体的过程,它能选择性地杀死染色体不稳定的癌细胞,而不会伤及其他细胞。该药物计划在2023年推向临床试验。我们正在探索的其他治疗策略包括:调节纺锤体的形成、改变细胞分裂期间染色体的组织,以及利用CIN驱动的炎症反应来抗癌。
寻找与CIN相关并且可作为治疗靶标的通路是件令人兴奋的事,它将打破部分癌症无药可及的局面。CIN是个很有吸引力的药物靶标,因为只有癌细胞会表现出染色体不稳定,那么针对CIN的治疗就不会错伤无辜——这是癌症治疗领域的圣杯。过去的十余年间,细胞生物学、基因组学及癌症生物学领域逐渐交织在一起,跨学科研究方法和学术界与产业界的合作将带来新的发现。这一切的终极目标是为病人服务——像我的那位肿瘤转移到脑部的病人,和其他现有的治疗选择十分有限的病人。
本文经授权编译自the-scientist.com,原标题为How Chaos in Chromosomes Helps Drive Cancer Spread阅读原文:https://www.the-scientist.com/features/how-chaos-in-chromosomes-helps-drive-cancer-spread-69695

0
推荐
 京公网安备 11010502034662号
京公网安备 11010502034662号