阅读:0
听报道
新冠疫情全球暴发,共享新型冠状病毒基因组序列十分重要。但在资源共享、推进科学发展对抗全球疫情的同时,也切不可放弃科学的严谨,警惕对于数据的片面放大解读。
文 | 继省 狄德罗 史隽
2020年2月28日,德国柏林Charité大学医院的病毒学家Christian Drosten发布了从一位德国患者身上分离出的新冠病毒 (SARS-CoV-2) 的基因组序列,随后,他立刻在twitter上发布了一条警告:目前,分析解读新冠病毒的基因序列还不是时候,很容易过度解读。随着病毒开始在全世界传播,全世界350多个科学家已经在GISAID (Global Initiative on Sharing All Influenza Data,一个旨在快速共享流感数据的网上平台) 平台上贡献了新冠基因组序列,这些信息为研究新冠病毒的传播和演化提供了线索。但是,这些序列仅占所有病例的极小一部分 (截止2020年3月10日,全球已经有近12万人感染了新冠病毒),而且其间只有很少的显著的序列差异,Drosten很快意识到,这些序列很容易被过度解读。

图1. 德国柏林Charité 大学医院的病毒学家Christian Drosten (来源于Berlin Institute of Health官网)
Drosten分析的新冠病毒基因序列来自于一位在意大利感染了新冠肺炎的德国人。这位患者的病毒基因组序列看起来和一位德国慕尼黑患者的类似,而这位慕尼黑的患者要比德国患者早一个多月感染新冠肺炎。两人的病毒基因组都有三个突变,是在早期中国患者的序列中没有发现的。Drosten意识到,这一发现可能会让人认为意大利的疫情暴发是由那位来自德国慕尼黑的病例“点燃”的。然而,巴伐利亚州 (慕尼黑是这个州的首府) 的公共卫生官员表示,经过追踪和隔离14个确诊病例的所有可能的接触者,巴伐利亚的疫情已被扑灭。Drosten认为,也不能排除另一种可能:携带有这三个基因突变的其他感染者分别接触了意大利和德国的患者。他在twitter上称,新公布的基因组“不足以表明慕尼黑和意大利的病例之间有关联”。
然而,Drosten的警告并没有引起学术界的重视。几天后,美国福瑞德·哈金森肿瘤研究中心 (Fred Hutchinson Cancer Research Center) 的Trevor Bedford分析了一系列的病毒基因组序列以后,连续发布推文进行讨论。他写道:根据基因组序列的分析,病毒传播的路线意味着德国巴伐利亚州的疫情根本没有得到遏制,并且很有可能导致了意大利暴发疫情。Bedford的言论传播得很广,《麻省理工技术评论》杂志断言,“慕尼黑病例可能与很大一部分的欧洲疫情有关联”,很多Twitter用户要求德国出来道歉。
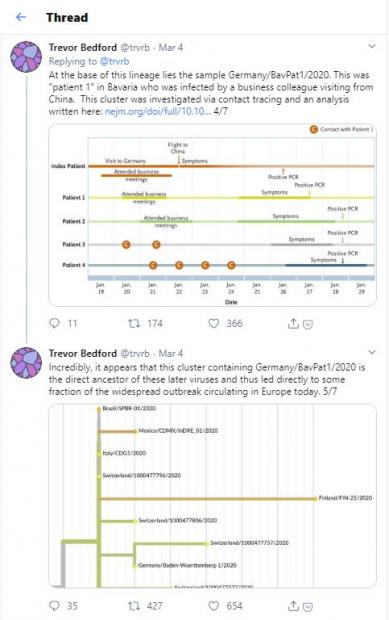
图2. Bedford发布的推文串(Threads)
欧洲疾控中心的病毒学家Eeva Broberg同意Drosten的观点。他也认为,比起还没有证明的巴伐利亚传播路线,还有很多可能的新冠病毒传播到意大利北部的路线。也有其他科学家认为Bedford话说得太早了些。其中一位与Bedford经常合作的瑞士巴塞尔大学(University of Basel) 的计算生物学家Richard Neher说:“我应该因为这个揍他两下。”爱丁堡大学的分子进化生物学家Andrew Rambaut说:“这件事警示了我。仅仅依据病毒谱系就断言病毒传播的路线是不对的”。Bedford后来也承认,不能排除从中国有两例分别传入意大利和德国的可能性。他认为自己发推文时应该“更加谨慎些”。
从上述这件事,我们可以看出在一个流行病暴发时,实时分析病毒基因组的优势,以及背后的陷阱。美国洛斯阿拉莫斯国家实验室 (Los Alamos National Laboratory) 的生物学家Bette Korber也在研究新冠病毒的基因组,他说:“新冠肺炎是一种非常重要的疾病。我们需要了解它的传播方式。在疫情暴发期间,病毒的进化非常有限,研究人员正在尽力分析,并提出建议,但是我认为眼下应当将这些说法视为建议,而不是定论。”
美国Scripps研究所的计算生物学家Kristian Andersen说,早期的基因序列数据是最重要的信息。2020年1月1日公布的第一个新冠病毒的基因序列就回答了最重要的问题:是什么病原体引起了新冠肺炎?随后公布的几个基因序列的相似度非常高,强烈暗示着病毒是由一个动物一次性地传染给了人类。如果新冠病毒是多次跨越物种壁垒传染的,科学家应该在最初的人类病例中看到更多的病毒变异。
现在,病毒变异逐渐涌现。和其他病毒一样,新冠病毒也时刻在发生随机突变;其中一部分会被病毒的纠错系统捕捉并修正。爱丁堡大学的分子进化生物学家Andrew Rambaut指出,新冠病毒的基因组有大约3万个碱基对,平均每个月就会累积出1-2个新的突变,这个速度比流感病毒慢2到4倍。利用这些细微的变化,研究人员就可以绘制出病毒的谱系树(phylogenetic tree,和人类的家谱类似)。他们也可以把新冠病毒病的案例都关联在一起,探究是否存在我们没监测到的病毒传播路径。
例如,当研究者对美国华盛顿州出现的第二株冠状病毒(来自2月27日确诊的一个少年)进行基因组测序后,发现它看起来像是第一株病毒(来自于6周前确诊的美国一号病人,一位从武汉探亲回美的35岁中国籍男子)的直接后代,并带有3个新的突变。Bedford在twitter上发表评论,认为这两个基因组来自不同的输入病例是“极不可能的”。他写道:“我相信我们正在面临的是,疫情在华盛顿州已经暴发,但是到现在还没有检测出来。”后来的情况证实这项研究的结论是正确的:华盛顿州现在已经报道了超过100例确诊和15例死亡,从这些病人身上分离的病毒的基因组也支持了第一二个病例之间的关联。Rambaut表示,这次Bedford的假说比上一个更可信,因为两例病人都来自华盛顿州的Snohomish县,而两株高度接近的病毒由不同途径分别准确传播到华盛顿州的同一个小城里,可能性是极低的。
目前,关于病毒传播途径的可靠结论还很少。—部分原因是,相比于全球10万以上的感染人数,被测试公布的病毒基因组只是九牛一毛。80%的新冠肺炎病例都在中国,但是在已公开的新冠病毒基因组里只有三分之一来自中国,并且来自于后期的病例的基因组极少。由于所采样品大多数来自暴发早期,绝大多数基因组非常相似,因此难以做出可靠结论。Neher说:“我们现在已知的病毒突变信息有限,分组分析都非常模棱两可。随着全世界的病例越来越多,我们预期能看到更多样的基因组和更清晰界定的病毒株系,到那时我们才能弄清真相。”
科学家们也在努力厘清基因组多样性背后的病毒突变,这些突变可能改变病毒致病力或传播速度。在这方面,同样需要谨慎下结论。北京大学Lu Jian等人3月3日在国家科学评论National Science Review上发表论文,分析了103例病毒基因组,发现这些病毒分属于两个亚型:S和L;70%的已测序新冠病毒的基因组都属于较新的L亚型。由此,Lu等人认为病毒通过演化变得更有致病性,传播更快。(详见《莫被误导!准确理解新冠病毒可能分为两种类型,且在暴发早期就已并存》)然而,Rambaut指出,Lu的分析缺乏证据,他们只是看到了病毒进化的两个分支,一个分支更大些,就下结论说这个分支的病毒一定更有致病性或更容易传播。但是,仅仅是病毒输出并在别处导致大暴发,并不代表病毒行为发生了改变;某个病毒株系比其他株系更庞大,完全有可能是偶然事件。已有研究者呼吁这篇论文撤稿。英国格拉斯哥大学(University of Glasgow)四位科学家联名在论坛(病毒学领域知名论坛,分析和解释病毒分子进化和流行病学)上发声,称这篇论文所得结论明显站不住脚,在病毒暴发如此关键的时期传播误导性信息非常危险。Lu则回复称四位同行误解了他们的研究。
Drosten说,多数基因组改变都不会改变病毒行为。要证实突变确实对病毒有影响,唯一的方式是,在细胞培养或动物模型中看到变异的病毒更容易进入细胞或传播。(见《传播力胜过SARS,需要担心新冠病毒变异吗?》)如果病毒确实发生了重大改变,也会有两种情况——危险性提高,或危险性降低。2018年Drosten课题组发表论文,他们发现,SARS冠状病毒在2002-2003年暴发早期,丢失了一小段基因组——某个基因上的29个碱基对。在实验室中把这些碱基对重新加回到病毒基因组里,病毒在多个细胞培养模型中都有更好的复制表现。这意味着SARS病毒在变异后危险性降低了。
弱化病毒的突变竟然会保留下来,可能看上去很诡异。但是当病毒刚进入人群传播,也不需要和没有这种突变的病毒竞争时,这种弱化突变存活的情况确实可能发生。Drosten说:“让人失望的是,新冠病毒并没有像SARS病毒一样出现弱化病毒的缺失。”
本文主要编译自
版权说明:欢迎个人转发,任何形式的媒体或机构未经授权,不得转载和摘编。转载授权请在「返朴」微信公众号内联系后台。
话题:

0
推荐
财新博客版权声明:财新博客所发布文章及图片之版权属博主本人及/或相关权利人所有,未经博主及/或相关权利人单独授权,任何网站、平面媒体不得予以转载。财新网对相关媒体的网站信息内容转载授权并不包括财新博客的文章及图片。博客文章均为作者个人观点,不代表财新网的立场和观点。
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}