阅读:0
听报道
本文要点:
1、抗体依赖增强(ADE)效应主要发生在具有Fc受体的免疫细胞。许多病毒(包括冠状病毒)都发现了ADE效应的证据,主要表现是增强病毒感染能力。
2、体外实验发现ADE现象,不代表一定会影响临床结果。
3、提高抗体质量是减少疫苗ADE风险的关键。
撰文 | Gene
近来,各国的新冠疫苗研发纷纷进入三期临床阶段,新冠病毒(SARS-CoV-2) 疫苗的安全性问题再次进入公众视野。不少文章都提到,ADE效应可能是新冠疫苗的潜在风险。
什么是ADE效应?ADE全称Antibody-dependent enhancement,意为抗体依赖性增强,比较通俗的解释是:病毒在感染细胞时,由于某些原因,体内已有的相关抗体会增强病毒的感染能力。换言之,经自然免疫或疫苗接种后,再次接触相关病毒时,体内产生的抗体可能会增强其感染能力,最终导致病情加重。
那么,ADE在科学上是如何解释的?新冠病毒是否也存在ADE效应?我们应该怎样避免?本文将深入介绍病毒的ADE效应,希望帮助大家正确理解科学现象和科学结论。
抗体依赖性增强效应的发现
抗体(antibody)最早是由德国科学家贝林(Emil Adolf Von Behring)和日本科学家北里柴三郎(Kitasato Shibasaburo)共同发现的。他们发现,将感染破伤风杆菌的兔子血清注入小鼠体内,可以使小鼠免受破伤风杆菌以及破伤风毒素的侵害[1]。随后,贝林又给豚鼠注射了灭活的白喉杆菌和白喉毒素,发现豚鼠的血清也具有了抗白喉杆菌和白喉毒素的保护性[2]。因此,贝林认为免疫后的动物血清中会产生一种名为“抗毒素(antitoxin)”的保护性物质,可以与外来抗原(antigen)反应而起作用。
“抗毒素”也就是后来所说的抗体,1891年,德国科学家埃尔利希(Paul Ehrlich)首次使用了“抗体”(antikörper)一词[3]。后来科学家又发现抗体主要分为五种亚型:IgA、IgD、IgE、IgG和IgM。
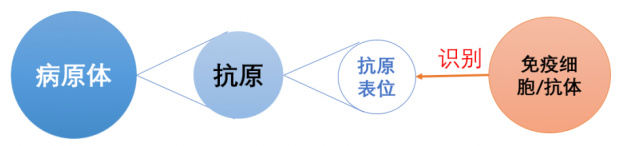
抗原(antigen)是指病原体上能够被免疫细胞特异性识别的分子。每个抗原可以有一个或多个抗原表位。抗原表位更加细致,它是抗原分子中决定抗原特异性的化学基团。免疫细胞(或抗体)主要通过识别抗原表位来与抗原相互作用,进而引发免疫反应。(见下图)
1964年,澳大利亚科学家Royle Hawkes在一次实验中意外地发现,在高度稀释的鸡抗体血清的环境中,黄病毒科的多种病毒对鸡胚成纤维细胞的感染性增强[4]。这一发现与“血清具有保护作用”的认识相矛盾,Hawkes对自己的发现产生了怀疑。
3年后,Hawkes终于证实,血清确实有可能增强病毒的感染性,并进一步发现,这一现象和血清中的IgG抗体有关[5]。抗体本是机体抵抗病毒入侵的盾牌,但病毒却可以“以子之盾,化己之矛”,依靠抗体的帮助入侵细胞。这是人类首次认识到病毒的抗体依赖性增强效应,但当时Hawkes并没能解释这一现象的具体机制。
现在,广义的ADE认为:
一些不理想的抗体可以增强病毒感染能力,甚至协助病毒进入原先无法进入的细胞,进而导致病毒大量复制或免疫细胞应答异常,最终使感染者病情加重,导致组织病理损伤。
直到 1977年,登革热领域的先驱、著名病毒学家Scott Halstead才将登革热病毒(dengue virus,DENV)在临床上引起的重症登革热和ADE联系起来——部分感染者康复之后获得了对登革热病毒的免疫力,然而一段时间后,当这些患者第二次感染登革热病毒时,病情反而比第一次更严重。
登革热病毒分为不同的血清型(即病毒的亚种),实验发现,对I型、III型和IV型具有免疫力的猴子在接受II型病毒感染后,体内的登革热病毒不但没有被清除,病毒水平反而还明显高于其他猴子。Halstead进一步发现,登革热病毒在具有免疫力的猴子或人的外周血白细胞中复制得更快。基于种种证据,Halstead得出结论,ADE和白细胞有关:在有抗体的条件下,病毒可以在白细胞中大量复制[6-8]。
为什么发生在白细胞?
这要从病毒感染细胞的步骤说起。病毒在进入人体后,首先通过自身的膜蛋白与人体细胞表面受体结合,之后通过膜融合或细胞内吞作用进入细胞,随后释放遗传物质,进行复制装配,最后释放病毒“子代”,继续感染其他细胞。
病毒入侵白细胞的过程亦不例外。Halstead解释说,ADE是由白细胞表面的Fc受体(FcR)介导发生的。在抗体的Fab段识别和结合病毒后,抗体的Fc段与白细胞(包括单核巨噬细胞、B细胞、中性粒细胞等)表面的Fc受体相互作用,使病毒粘附于白细胞表面,促进了白细胞对病毒的内吞作用,相当于“引狼入室”,增强了病毒的感染能力。这也是目前ADE发生的最主要机制。
什么是抗体的Fab段和Fc段?一张图带你认识——
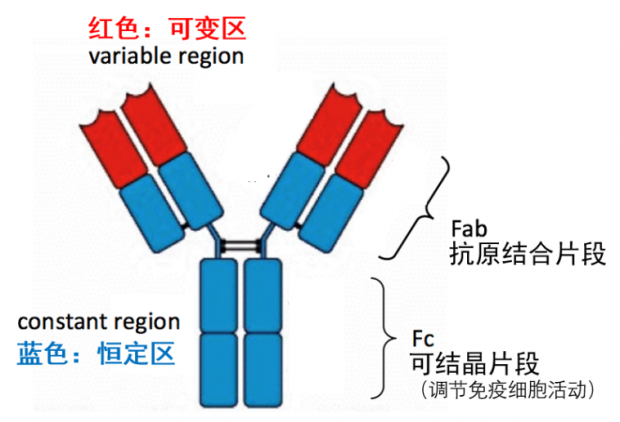
图1. 抗体即免疫球蛋白(immunoglobulin, Ig)分子,基本结构呈“Y”字形。Y字形的两臂是识别外来抗原的关键所在,所以也称为抗原结合片段(fragment antigen binding),即Fab 段;Y字形的根部称为可结晶片段(fragment crystallizable),即Fc段,主要负责调节免疫细胞活动。另外,Fc段也与ADE有关。| 作者作图
随后,著名病毒学家、香港大学公共卫生学院前院长Malik Peiris通过更详细的实验证据阐明了这一机制[9, 10]。Peiris发现,在西尼罗病毒(WNV,属于黄病毒科)感染巨噬细胞系的过程中,阻断白细胞表面的特定Fc受体与抗体Fc段的结合,就可以阻断病毒感染的ADE效应。其他研究者在登革热病毒和黄热病毒(YFV,属于黄病毒科)的实验中也得到了同样的结论[11, 12]。黄病毒科因为ADE而一时名声大噪。
ADE机制不止一种
1983年,马来西亚病毒学家Jane Cardosa发现了黄病毒科的另一种ADE机制。实验中,在IgM抗体存在的条件下,西尼罗病毒对淋巴瘤细胞的感染性增强。然而,像过去一样,阻断细胞表面的Fc受体,却不再有用;而如果阻断抗体Fc段与细胞表面的III型补体受体(CR3)的结合,则可以停止病毒的感染性增强作用[13]。
补体(complement)是血清中一组活化后具有生物活性的蛋白质,可对特异性抗体起到补充和辅助作用,主要介导非特异性免疫和炎症反应。补体系统包括补体固有成分、补体调控成分和补体受体(CR)。
这意味着,Cardosa实验中出现的ADE效应是由细胞表面的补体受体介导的。IgM抗体的Fab段识别并结合病毒后,抗体的构象改变,暴露出Fc段的补体结合位点——本来,这是为了激活补体系统,帮助抵抗病毒,然而出招便露破绽——补体系统被激活后,病毒-抗体复合物与靶细胞上的补体受体相结合,反把病毒送进了细胞内部,进一步增强了感染。
这一途径独立于Fc受体介导的ADE,因为Fc受体只在免疫细胞中表达,而补体受体表达的细胞类型则相对较广[14],病毒加剧入侵的细胞范围也更广。
目前,Fc受体介导和补体受体介导是ADE最常见的两种机制。除了黄病毒科外,科学家们也相继在其他病毒科的多种病毒中都发现了ADE现象,其中机制亦不完全相同。
冠状病毒中的ADE效应
冠状病毒(CoV)中的ADE效应首次发现于1980年[15]。著名冠状病毒学家Niels Petersen对幼猫进行猫冠状病毒的感染实验,引发猫传染性腹膜炎(FIP)。实验中,他发现自然条件下猫传腹病毒FIPV*抗体阳性的幼猫比抗体阴性的幼猫发病时间更早,死亡也更快,也就是说对FIPV有免疫力的幼猫在被感染后,疾病反而更严重。
*注:FIPV是猫冠状病毒FCoV的一种。
一年后,研究者证实,预先注射抗FIPV血清或抗体(实验中称为被动免疫)的幼猫,在感染FIPV时,发病时间和死亡时间同样早于对照组的幼猫[16]。1990年,研究者给幼猫打FIPV疫苗(实验中称为主动免疫),在体内确认检测到抗体后,再用FIPV去感染这些幼猫,也得到了相同的结果[17]。至此,FIPV感染过程中的ADE现象终于广为人知。
又过了两年,研究者才发现了猫冠状病毒ADE效应的机制。原来,某些抗FIPV的IgG抗体可以增强FIPV对巨噬细胞的感染能力,且该过程和 Fc受体相关[18]。此后,对FIPV 的ADE效应的研究越来越多。

图2. Petersen与FIPV感染康复的小猫Tony[19]。
2005年,研究人员在实验中首次发现,针对人SARS冠状病毒(SARS-CoV)的抗体可以增强另一种SARS毒株对宿主细胞的感染[20],并且在人B细胞和巨噬细胞中,SARS病毒的ADE效应与特定类型的Fc受体(FcγRII)相关,阻断这一受体可以阻断ADE的发生[21, 22]。
值得注意的是,SARS-CoV通过ADE感染巨噬细胞的过程,并非是通过单纯大量复制病毒加剧感染(图3A),而是干扰各种细胞因子信号(图3B),导致巨噬细胞在中后期负担过重,出现活化异常,炎症因子分泌增加,最终造成急性炎症和机体病理损伤[23, 24]。
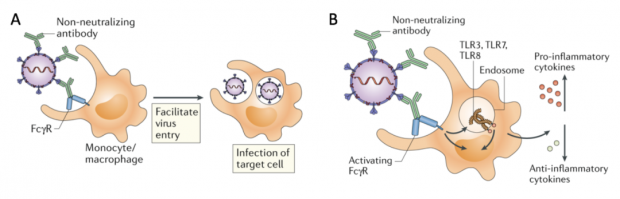
图3. 不理想的抗体导致冠状病毒感染加剧的两种方式。绿色代表抗体,黄色代表细胞,细胞表面突起的蓝色为Fc受体。| 改编自参考文献[25]。
另一项针对MERS冠状病毒(MERS-CoV)感染的ADE的体外研究发现,有些不理想的抗体和病毒表面的刺突蛋白结合后,可以使刺突蛋白的构象发生改变,结果,不但病毒仍然可以和相应的细胞表面受体结合,同时抗体的Fc段也可以与细胞表面的Fc受体结合,反而让病毒更容易进入细胞[26]。这说明如果初次感染时诱导的抗体不够理想,也可能直接引发ADE效应。
基于SARS 和MERS冠状病毒的证据以及临床研究,已有研究者合理推测,新型冠状病毒SARS-CoV-2感染也存在ADE效应[27, 28]。最近的一项体外研究(预印本)显示,SARS-CoV-2的单克隆抗体MW05可能通过Fc段与靶细胞表面的特定受体(FcγRIIB)结合,引起ADE效应,具体结果仍需进一步验证[29]。除此之外,另一项预印本研究显示,在新冠病毒感染的重症患者中,IgG抗体可能会诱导巨噬细胞产生超炎症反应,进而损坏肺内皮细胞屏障的完整性,引发微血管血栓[30]。
什么是“不理想”的抗体?
决定抗体是否会引起ADE的因素主要包括:抗体的特异性、滴度、亲和力以及抗体的亚型[25]。
SARS 疫苗包括不同种类,针对刺突蛋白(S蛋白)的疫苗和核衣壳蛋白(N蛋白)的疫苗所选择的抗原不同,诱导出的特异性抗体也不同。在小鼠实验中,给小鼠打疫苗后, 这两类疫苗诱导出的特异性抗体滴度是相似的,随后,再让这些小鼠感染SARS-CoV,发现编码N蛋白的疫苗会诱导小鼠分泌更多的促炎症因子,小鼠体内某些白细胞的肺部渗透也相对增加,肺病理学变化相对更为严重[31]。
相似地,在猴子模型中,针对SARS-CoV的刺突蛋白的不同表位的抗体,其诱导的反应也各不相同,有些可以起到很好的保护作用,有些则容易引起ADE效应[32]。
抗体滴度低也容易引起ADE效应,例如在SARS或MERS冠状病毒感染过程中,如果增加抗体滴度则可以抑制ADE,并促进中和反应的发生[26, 33]。在中和反应过程中,高亲和力的抗体还会比低亲和力的抗体保护效果更好[34]。
具有中和作用的抗体叫做中和性抗体。中和作用指的是抗体Fab段与相应的抗原表位结合,封阻其受体结合位点或致其构象改变,使抗原无法进入细胞。
抗体的亲和力,通俗来讲是指抗体同抗原结合的牢固程度。
此外,抗体的亚型不同,其Fc段调节免疫细胞的功能也各不相同:IgM能够更有效的激活补体系统,产生促炎症反应,IgG则根据细胞表面不同的Fc受体来调节免疫反应,如在SARS-CoV感染过程中,有些类型的Fc受体(FcγRIIa和FcγRIIb)可以介导ADE发生,有些(FcγRI 和FcγRIIIa)则不能[33]。进一步的,同一类型的Fc受体的不同剪接体(isoform),引发的ADE效应也不尽相同[35]。
疫苗研发如何避免ADE?
在新冠疫苗的研发过程中,减少ADE风险的关键在于提高抗体的质量,主要包括抗原表位与佐剂的选择。
抗原表位的选择尤其重要。此前SARS疫苗的开发过程中,在小鼠或猴子身上,有些疫苗一定程度上可以引发ADE效应,或引起由嗜酸性粒细胞介导的免疫病理学变化[20, 23, 36]。究其原因,可能是疫苗中起主要贡献的优势抗原表位诱导出的抗体质量(主要是滴度)不理想。
所谓佐剂,就是预先或与抗原同时注射的物质。佐剂可以有效增强机体对抗原的免疫应答,也可以改变免疫反应类型。研究显示,在老年小鼠中,铝佐剂增强的灭活SARS疫苗可以诱导出高滴度的抗体,但却是不理想的抗体亚型。此外,不适当的佐剂还会改变免疫反应类型,进而影响免疫应答过程,引起肺病理学变化[36]。
除此之外,疫苗的接种途径也会影响其作用。针对同一种SARS 疫苗,分别经鼻腔途径或肌肉途径接种,再经病毒感染后,前一种途径的接种者出现的肺部病理学变化更少[37]。另外也有研究显示,利用生物手段为疫苗颗粒表面包装一层外壳,例如在登革热疫苗颗粒表面包装磷酸钙矿化外壳,可以在不影响其保护效果的同时,有效避免ADE现象的发生[38]。
从ADE的发生机制入手,也能为疫苗研发“避雷”。既然大部分ADE效应是由细胞表面 Fc受体介导发生的,那么封阻细胞表面的特定Fc受体,则可以防止病毒-抗体复合物与Fc受体结合,进而阻止ADE效应[39]。
要想实现这一过程,针对Fc受体的特异性抗体,或抑制结合过程的小分子抑制剂都是不错的选择,前者可以作为免疫抑制剂使用[40,41]。例如,临床上对重症COVID-19患者使用静脉注射免疫球蛋白,可以改善患者症状[42, 43],但大范围是否安全有效还需进一步研究。
总之,通过封阻病毒-抗体复合物与Fc受体结合也是一种阻止ADE发生的手段,但是除了Fc受体外,ADE仍可以通过前述其他途径,如补体介导发生。
因此,在开发疫苗时,不但要保证诱导出高质量的中和抗体,最重要的是还要尽量选择可以诱导强细胞免疫的疫苗。实际上,机体清除病毒也依赖于细胞免疫,因为中和抗体只能对细胞外的病毒起作用,对于进入细胞内的“漏网之鱼”往往无能为力。病毒在细胞内会将其蛋白信息表达在感染的细胞表面,而杀伤性T细胞能够识别这些信息,从而发动攻击,将病毒与其感染的细胞共同杀灭。
同样重要的是,初次免疫(即疫苗接种)除了诱导出抗体外,还会产生记忆细胞。疫苗诱导的细胞免疫越强,激活的杀伤性T细胞就越多,转化的记忆性T细胞就越多,这样一来,在下次病毒感染时,免疫细胞行使功能的速度也就越快,从而有效地减少ADE的发生。因此,疫苗的种类选择也至关重要。
结 语
新冠疫苗研发至今,已公布的多项动物结果和临床试验结果均未出现明确的ADE证据。但是基于SARS疫苗和MERS疫苗的经验,笔者认为,在极个别新冠病毒单克隆抗体中发现ADE效应的确证,大概率只是时间问题。
虽然前文写到,已有研究初步表明新冠病毒的某些单克隆抗体在体外可能存在ADE效应,但目前证据仍不充分。更需要注意的是,体外实验与体内情况往往有较大差距,离临床表现更是相去甚远。机体经抗原免疫后,会出现针对多个表位的多克隆抗体反应,即便单个抗体具有ADE效应,也难以影响血清的中和性。
单克隆抗体是由单一B细胞克隆产生的、仅针对某一特定抗原表位的抗体。相应的,多克隆抗体是针对多种抗原表位的不同抗体。
另外,除了疫苗以外,开发单克隆抗体、制备抗体药物也是一种不错的选择。单克隆抗体具有分子精度,易于通过基因工程学编辑,如仅使用抗体的Fab段、或使用工程学对抗体的Fc段进行改造(如引入突变),都可以显著提高安全性[44]。
目前,世界各地的科研团队正在开发的新冠疫苗已有百种,其中至少30种已进入临床试验阶段(中国有10种),最快的已经开展临床III期,其余还有多种正在动物模型上开展试验[45]。同时,单克隆抗体的开发竞赛也正如火如荼。笔者认为,ADE不会成为新冠疫苗开发过程中的障碍。
参考文献
[1] Behring E, Kitasato S. Über das Zustandekommen der Diphtherie-Immunität und der Tetanus-Immunität bei Thieren. Dtsch Med Wochenschrift 1890; 49:1113–1114.
[2] Behring E. Untersuchungen ueber das Zustandekommen der Diphtherie-Immunität bei Thieren. Dtsch Med Wochenschrift. 1890; 50:1145–1148.
[3] Lindenmann J. Origin of the terms 'antibody' and 'antigen'. Scand J Immunol. 1984 Apr;19(4):281-5.
[4] Hawkes RA. Enhancement of the infectivity of arboviruses by specific antisera produced in domestic fowls. Aust J Exp Biol Med Sci 1964; 42: 465–482.
[5] Hawkes RA, Lafferty KJ. The enhancement of virus infectivity by antibody. Virology 1967; 33: 250–261.
[6] Halstead SB, O’Rourke EJ. Antibody-enhanced dengue virus infection in primate leukocyte. Nature 1977; 265: 739–741.
[7] Halstead SB, O’Rourke EJ. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J Exp Med 1977; 146: 201–217.
[8] Halstead SB, O’Rourke EJ, Allison AC. Dengue viruses and mononuclear phagocytes. II. Identity of blood and tissue leukocytes supporting in vitro infection. J Exp Med 1977; 146: 218–229.
[9] Peiris JS, Porterfield JS. Antibody-mediated enhancement of flavivirus replication in macrophage-like cell lines. Nature 1979; 282: 509–511.
[10] Peiris JS., et al. Monoclonal anti-Fc receptor IgG blocks antibody enhancement of viral replication in macrophages. Nature 1981; 289: 189–191.
[11] Daughaday CC., et al. Evidence for two mechanisms of dengue virus infection of adherent human monocytes: trypsin-sensitive virus receptors and trypsinresistant immune complex receptors. Infect Immun 1981; 32: 469–473.
[12] Schlesinger JJ, Brandriss MW. Antibody-mediated infection of macrophages and macrophage-like cell lines with 17D-yellow fever virus. J Med Virol 1981; 8: 103–117.
[13] Cardosa MJ., et al. Complement receptor mediates enhanced flavivirus replication in macrophages. J Exp Med 1983; 158: 258–263.
[14] Ross GD. Complement receptors. In Encyclopedia of Immunology, Roitt IM, Delves PJ (eds). Academic Press: San Diego, 1992; 388–391.
[15] Petersen, N.C. and J.F. Boyle. Immunologic phenomena in the effusive form of feline infectious peritonitis. Am J Vet Res 1980; 41:868–876.
[16] Weiss, R.C. and F.W. Scott. Antibody-mediated enhancement of disease in feline infectious peritonitis: comparisons with dengue hemorrhagic fever. Comp Immunol Microbiol Infect Dis 1981; 4:175–189.
[17] Vennema, H., et al. Early death after feline infectious peritonitis virus challenge due to recombinant vaccinia virus immunization. J Virol 1990; 64:1407–1409.
[18] Olsen C.W., et al. Monoclonal antibodies to the spike protein of feline infectious peritonitis virus mediate antibody-dependent enhancement of infection of feline macrophages. J Virol 1992; 66:956–965.
[19]
[20] Yang ZY., et al. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc Natl Acad Sci U S A. 2005 Jan 18; 102(3): 797–801.
[21] Kam YW., et al. Antibodies against trimeric S glycoprotein protect hamsters against SARS-CoV challenge despite their capacity to mediate FcγRII-dependent entry into B cells in vitro. Vaccine. 2007 Jan 8;25(4):729-40. Epub 2006 Aug 22.
[22] Yip MS., Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol J. 2014 May 6;11:82. doi: 10.1186/1743-422X-11-82.
[23] Liu L., et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 2019;4, e123158.
[24] Yip MS., et al Antibody-dependent enhancement of SARS coronavirus infection and its role in the pathogenesis of SARS Hong Kong Med J 2016;22(Suppl 4):S25-31
[25.] Iwasaki A., et al. The potential danger of suboptimal antibody responses in COVID-19. Nat Rev Immunol. 2020 Apr 21.
[26] Wan Y., et al. Molecular Mechanism for Antibody-Dependent Enhancement of Coronavirus Entry. J Virol. 2020 Feb 14;94(5). pii: e02015-19.
[27] Tetro JA. Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect. 2020 Mar;22(2):72-73.
[28] Li H., et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. 2020 Apr 17. pii: S0140-6736(20)30920-X.
[29] Wang S., et al. An antibody-dependent enhancement (ADE) activity eliminated neutralizing antibody with potent prophylactic and therapeutic efficacy against SARS-CoV-2 in rhesus monkeys. bioRxiv. Posted July 27, 2020.
[30] Hoepel W., et al. Anti-SARS-CoV-2 IgG from severely ill COVID-19 patients promotes macrophage hyper-inflammatory responses. bioRxiv. Posted July 13, 2020.
[31] Yasui F., et al. Prior immunization with severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV) nucleocapsid protein causes severe pneumonia in mice infected with SARS-CoV. J. Immunol. 2008;181, 6337–6348.
[32] Wang Q., et al. Immunodominant SARS coronavirus epitopes in humans elicited both enhancing and neutralizing effects on infection in non-human primates. ACS Infect. Dis. 2016;2, 361–376.
[33] Li H., et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. 2020 Apr 17. pii: S0140-6736(20)30920-X.
[34] Pierson TC., et al. Structural insights into the mechanisms of antibody-mediated neutralization of flavivirus infection: implications for vaccine development. Cell Host Microbe 2008;4, 229–238.
[35] Yuan FF., et al. Influence of FcγRIIA and MBL polymorphisms on severe acute respiratory syndrome. Tissue Antigens 2005;66, 291–296.
[36] Bolles M., et al. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J. Virol. 2011;85, 12201–12215.
[37] Du L., et al. The spike protein of SARS-CoV — a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009;7, 226–236.
[48] Wang X., et al. Biomimetic inorganic camouflage circumvents antibody-dependent enhancement of infection. Chem Sci. 2017 Dec 1;8(12):8240-8246.
[49] Fu Y., et al. Understanding SARS-CoV-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol Sin. 2020 Mar 3.
[40] van Mirre E., et al. IVIg-mediated amelioration of murine ITP via FcgammaRIIb is not necessarily independent of SHIP-1 and SHP-1 activity. Blood. 2004 Mar 1;103(5):1973; author reply 1974.
[41] Veri MC., et al. Monoclonal antibodies capable of discriminating the human inhibitory Fcgamma-receptor IIB (CD32B) from the activating Fcgamma-receptor IIA (CD32A): biochemical, biological and functional characterization. Immunology. 2007 Jul;121(3):392-404.
[42] Cao W., et al. High- Dose Intravenous Immunoglobulin as a Therapeutic Option for Deteriorating Patients With Coronavirus Disease 2019. Open Forum Infectious Diseases 2020;7.
[43] Shao Z., et al. Clinical efficacy of intravenous immunoglobulin therapy in critical patients with COVID-19: A multicenter retrospective cohort study. medRxiv. Posted April 20, 2020.
[44.] 57. Amanat F, Krammer F. SARS-CoV-2 Vaccines: Status Report. Immunity. 2020 Apr 14;52(4):583-589.
[45] Le TT., et al. Evolution of the COVID-19 vaccine development landscape. Nat Rev Drug Discov. 2020 Sep 4. doi: 10.1038/d41573-020-00151-8.
话题:

0
推荐
财新博客版权声明:财新博客所发布文章及图片之版权属博主本人及/或相关权利人所有,未经博主及/或相关权利人单独授权,任何网站、平面媒体不得予以转载。财新网对相关媒体的网站信息内容转载授权并不包括财新博客的文章及图片。博客文章均为作者个人观点,不代表财新网的立场和观点。
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}