撰文 | 刘翔宇
GPCR简介
想象一个美好的周五傍晚,你终于完成了一周辛苦的工作,看了一眼窗外华灯初上,推开窗闻到了一阵食物的芬芳,你觉得饿了。你出门想找点吃的,路上一辆摩托车贴着你开过,吓得你心跳快了半拍。终于到了饭店,你点了烤串和啤酒,咬一口滋滋作响的羊肉,油脂香混合着幸福感直冲大脑,你感到一阵快乐……
在这个事件中,G蛋白偶联受体(简称GPCR)介导几乎每一步反应。视紫红质的激活让你看到东西,嗅觉受体的激活让你闻到香味,饥饿素受体的激活让你想摄入食物,肾上腺素受体的激活让你心跳加快,味觉受体的激活让你感知食物的美味,多巴胺受体的激活让你感到快乐。

图1:GPCR调控人体的行为。如食欲、睡眠、瘙痒、应激反应等一系列的生理过程均由GPCR介导调控。丨图画作者:张欣
GPCR是一类具有七次跨膜螺旋的膜蛋白受体,在人体里面有超过800个家族成员,它们参与调节人类生命活动的方方面面(图1)。如果人类是由化学反应调控的机器的话,GPCR就是机器里面的调节按钮。如果我们通过药物来调控这些按钮GPCR的活性,就可以调节人的生理病理状态,从而起到治疗疾病的目的。因为这个原因,GPCR是非常重要的药物靶点,目前FDA批准的药物约三分之一通过靶向GPCR来发挥药效[1]。在我开始GPCR研究之后,我就养成了仔细阅读药品说明书的习惯。有一次我因为咽炎咳嗽,大夫给我开了“开瑞坦”和“孟鲁斯特纳”,我一读说明书发现前者是组胺(H1)受体拮抗剂,后者是白三烯受体(CysLR1)拮抗剂,越发让我坚信研究GPCR大有可为。此外还有很多GPCR药物用于治疗多种威胁生命的重大疾病,如β肾上腺素受体阻滞剂美替洛尔(Metipranolol)用于治疗心律不齐和高血压;而β2肾上腺素受体激动剂福莫特罗(Formoterol)和沙美特罗(Salmeterol)用于治疗慢阻肺和哮喘;5-羟色胺受体1A的激动剂阿立哌唑(Aripiprazole)和维拉唑酮(Vilazodone)用于治疗精神分裂症和中重度抑郁症等。众多GPCR靶向的药物及其庞大的市场份额(约占全部治疗药物市场份额的27%)也反映了GPCR作为药物靶点的潜能与重大意义。
GPCR结构解析历史
由于GPCR重要的生理和药理功能,解析其结构成为了许多结构生物学家的目标。在这个过程中不少科学家采用不同的方法做出了杰出的工作,其中尤为突出的是一个半路出家进入结构领域的医学博士——斯坦福大学的Brian Kobilka教授。我将主要介绍Brian的工作,一方面因为Brian在这个方向做的工作尤其出色,另一方面因为Brian是我的博士后导师,我对他的工作比较熟悉。
在人的GPCR结构被解析之前,两类七次跨膜的膜蛋白结构被解析。一类是来自盐沼盐杆菌(Halobacterium salinarium)的细菌视紫红质(Bacteriorhodopsin)[2-5],一类是牛眼睛里面提出来的视紫红质(Rhodopsin) [6]。其中细菌视紫红质虽然是七次跨膜蛋白,但它是一类感光的离子通道,工作机理不同于G蛋白偶联受体,结构上也和GPCR略有差异。牛眼睛里提出的视紫红质结构是第一个G蛋白偶联受体结构[6]。视紫红质的结构工作对于后续其他GPCR的结构解析提供了指导。
相比于视紫红质的结构解析工作,β2AR(β2肾上腺素受体)的结构解析有几个额外的难点。第一是如何获得大量的蛋白。视紫红质可以大量从牛眼睛里获得,而β2AR在天然组织里的丰度很低,从天然组织纯化需要大量的工作。在上个世纪70年代中期杜克大学Robert Lefkowitz教授实验室曾经开展过从天然组织里大量纯化β2AR的工作。他们需要每周从大概2000只青蛙的血红细胞里面提β2AR,而其实验室最多的时候也拿不到超过五十微克的β2AR[7]。晶体结构解析需要毫克级别的蛋白,因此研究者们需要开发异源重组表达和纯化β2AR的方法。在1995年Brian曾发表过一篇单独作者的Analytical Biochemistry文章,讲述怎么在β2AR的氨基端和羧基端分别加上亲和标签,帮助纯化利用昆虫细胞表达出来的β2AR,这样可以拿到毫克级别的受体[8]。这个方法我们直到今天还在实验室里面使用。
解析β2AR结构的第二个难点在于如何稳定受体的构象。晶体结构解析需要足够纯的蛋白,这个“纯”不仅指没有其他的杂蛋白,还指同样的蛋白要维持在相同的构象。而GPCR作为一个受体蛋白,它的功能就是感知信号,传递信号。为了实现这个功能,受体的结构要足够灵活多变——这个特点和晶体结构解析正好矛盾。想象一下给一个调皮捣蛋上蹿下跳的孩子拍一张不糊的照片有多难,解析一个状态多变的蛋白的晶体结构更要难上百倍。视紫红质虽然也是GPCR,但它没有基础活性 (basal activity)。这可以理解,没有光的时候我们什么都看不到。而β2AR是有基础活性的,即便没有肾上腺素去激活它,它也会传递一些信号。视紫红质的配体是共价连在受体上的,而β2AR的配体是自由扩散的,这也意味着可能存在一些受体上结合了配体,一些没有结合的情况。这种情况也会严重影响晶体的生长。为了解决这个问题需要使用一些具有很高亲和力的配体。好在肾上腺素受体的配体是心脏和呼吸道疾病的潜在药物,吸引了众多研究者包括制药公司在内的广泛兴趣,因此存在大量具有不同性质的β2AR配体可供选择。值得一提的是为了将GPCR稳定在特定构象,剑桥大学Christ Tate教授实验室曾采用丙氨酸扫描(alanine scan)的方法,对火鸡的β1AR的每一个氨基酸都进行点突变研究突变体的性质,最终得到热稳定性提升的β1AR突变体[9]并解析了结构[10]。这种引入热稳定突变的方法也后续帮助解析了不少GPCR结构。
解析β2AR结构的第三个难点和蛋白晶体生长的原理相关。所谓的蛋白质晶体是由一个一个蛋白质分子堆积起来的。想象一下国庆阅兵时整齐排列的方队,那可以说是一个二维晶体,把这样的二维晶体在三维规则排列起来就组成了三维晶体。而在晶体里蛋白质分子和分子之间是需要相互作用的,这和阅兵方阵不一样,除非方阵里面的每个人和边上的人手拉手互相连起来。GPCR是一类跨膜蛋白,为了把它们从细胞膜上溶下来,纯化时需要加上去垢剂。GPCR的跨膜区被去垢剂微团(detergent micelle)包起来,就像在一些综艺节目里面人钻进了充气的气球里面,只有短短的手露在外面。这种情况下手拉手就很困难,所以需要增加露出去垢剂微团的部分的大小。在GPCR晶体工作里面,专业的说法是增加可溶区的面积。Brian和合作者在这里采用了两种不同的方法,一是采用了一个特异性识别受体的抗体可变区(Fab)[11];另一个是在β2AR的第五个和第六个跨膜螺旋之间融合了一个很容易结晶的溶菌酶[12, 13](图2)。这两个方法都成功了!尤其是后一个方法后来成为了解析GPCR晶体结构的通用方法。我对融合溶菌酶这个方法一直抱着“这也能行?!”的态度。因为我从自己学习和研究蛋白结构的角度来看,融合溶菌酶的方法成功率应该很低:如何将两个蛋白直接融合在一起,让它们之间有足够的距离,这样彼此不挤着对方;又让它们足够靠近,这样二者之间可以保持比较固定的相对位置。因为如果融合上的溶菌酶相对于β2AR左右乱摆的话,融合蛋白的构象就只会更乱,更加不利于结晶。但是Brian实验室就是这么尝试并且成功了!通过这个故事我也一直提醒自己:在研究过程中有一些规律或者经验是不可靠的,不要让这些不可靠的经验限制了对未知世界的探索。不过这个领悟就像就像所有的其他道理一样:知易行难。虽然懂了这个道理,在实际操作时往往会忘记。
通过解决这一系列的问题,Brian实验室与合作者一起在2007年连续发表了多篇β2AR的晶体结构文章(图2)[11-13]。但是这些结构捕捉的都是β2AR结合拮抗剂的非激活状态。为了解释β2AR如何在结合激动剂之后被激活,进而将信号传递给G蛋白,Brian实验室又尝试解析β2AR的激活态的结构。那一系列的工作真是精彩绝伦。这里我就不过多描述了,如果感兴趣可以去阅读一下他们实验室在2011年发表的β2AR-Gs复合物结构的文章14。在这个工作里面整合了多个研究组最领先的技术:1、利用了β2AR的超强激动剂;2、利用核苷酸酶去除系统里面的GDP,将复合物诱导在相对稳定的无核酸结合状态;3、纯化过程使用了新开发的去垢剂LMNG(后来成为了GPCR结构研究最成功的去垢剂);4、利用氨基端融合的T4溶菌酶增加晶体堆积的可能性;5、开发纳米抗体来稳定β2AR-Gs复合物;6、利用了Martin Caffery实验室最新开发的新型脂质MAG7.7来作为晶体生长的host lipid。再加上Brian实验室和Roger Sunahara实验室多年来对制备GPCR和G蛋白的条件摸索和经验积累。这个工作实在是令人叹为观止,我至今记得第一次知道这个工作时被震撼的感觉。四年之后,当我看到徐华强老师课题组解析的rhodopsin-arrestin复合物结构[15]时才有类似的感觉。

图2:Brian Kobilka实验室在2007年到2011年间解析的部分非激活态(绿色)和激活态(橙色)的β2AR结构。每个结构的下标为该结构的PDB号以及该结构发表的时间。
GPCR结构如何指导药物开发
随着受体表达纯化条件的成熟以及电镜技术的革命性突破,目前已经有许多不同GPCR结合不同配体的激活态或非激活态结构被报道出来。这些结构大大增进了我们对药物与GPCR相互作用和GPCR与下游G蛋白或者arrestin信号传递分子机理的理解。GPCR一直备受关注的主要原因是因为它们是重要的药物靶点。在获得了这么多的结构信息之后,这些信息如何能够指导我们设计更好的药物呢?我将利用下面三个例子加以介绍。
第一个例子是利用结构指导药物的改造。这是由我博士后时的实验室(清华大学Kobilka实验室)和合作者们一起完成的一个课题。在这个工作里我们试图开发一种对M3乙酰胆碱受体具有选择性的抑制剂,这样的化合物可能对如慢阻肺等呼吸道疾病具有治疗效果。而我们想减少对M2乙酰胆碱受体的抑制活性,以尽量减少对心脏的潜在副作用。这个工作的难点在于M2受体和M3受体的正构配体结合口袋非常相似,几乎一模一样。只有一个氨基酸不同。这个氨基酸在M2受体中是比较大的苯丙氨酸(Phe),在M3受体中是比较小的亮氨酸(Leu)。针对这单个氨基酸的区别,我们通过结构分析、分子对接、动态模拟、化学合成等手段,改造了一个本来对M3受体没有选择性的药物。改造过程中,在化合物的一个特定位置引入一个氟原子。按照基于结构的设计,这个氟原子可以和M3受体中比较小的亮氨酸共存,但是却会和M2受体中比较大的苯丙氨酸在位置上有冲突。通过这种方式,我们得到了具有M3受体选择性(体外实验105倍、体内实验>1000倍)的小分子化合物并利用结构生物学验证了其选择性的分子基础[16]。这个工作证明了基于结构精准改造药物的可行性。
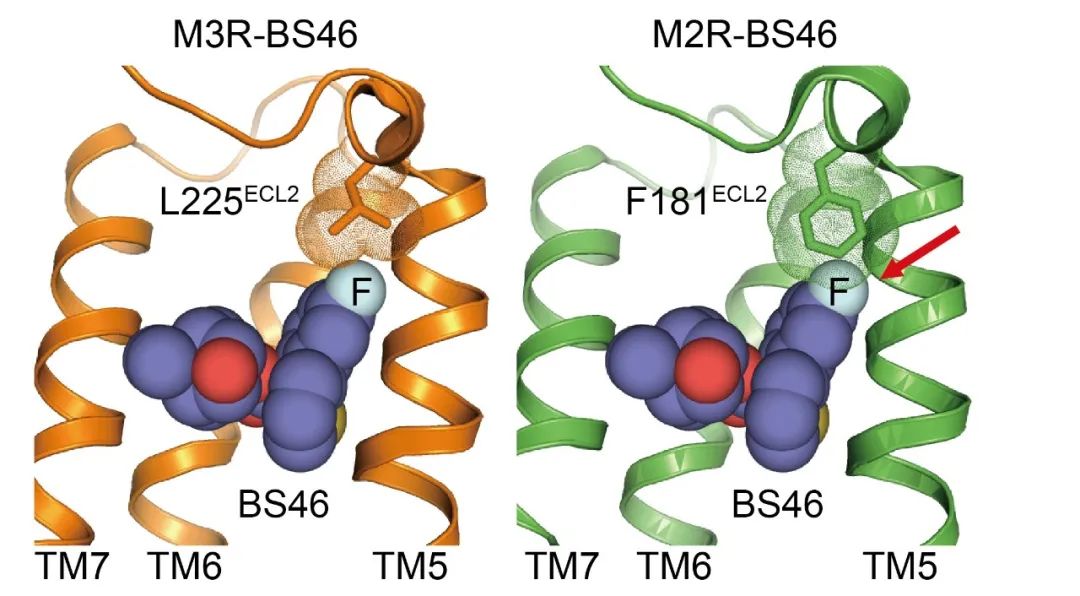
图3:改造后的M3R选择性配体BS46结合M3受体(橙色)时氟原子(天蓝色)可以和亮氨酸(L225)共存。如果把BS46放进M2受体(绿色)的结合口袋里面,氟原子会和苯丙氨酸(F181)发生位置冲突(红色箭头指示处)。利用这个设计,BS46实现了对M3受体的选择性[16]。
第二个例子是利用结构进行分子对接,虚拟筛选目的GPCR的药物分子。这是来自UCSF大学的Brian Schoichet教授和John Irwin教授与北卡罗来纳州大学教堂山分校Bryan Roth教授的合作成果。在这个工作里,研究者们针对D4多巴胺受体(精神分裂症和帕金森症的药物靶标)进行了一个大规模的虚拟筛选。他们一共对D4多巴胺受体的正构配体结合口袋虚拟对接了1.38亿种不同的化合物。由于每种化合物有不同的对接模式,他们一共需要计算70万亿(70,000,000,000,000)种不同的结合方式。这个数字听起来特别庞大,但是利用强大的计算资源,在这个课题中他们只花了不到一天半就算完了。注意这是2019年发表的工作,随着计算能力的进一步提升,将来需要的时间会更短。在他们虚拟筛选出的化合物中,有一个化合物(ZINC621433143)表现出2.3 nM的高亲和力。这个化合物是多个手性异构体的混合物。它的其中一个手性异构体(ZINC621433144)表现出对D4多巴胺受体0.18 nM的亲和力,以及相比于其他亚型的多巴胺受体超过2500倍的选择性。这样,他们通过虚拟筛选直接找到了目前对D4多巴胺受体亲和力最高,选择性最强的激动剂。这样的化合物可能具有治疗精神类疾病的潜力。
第三个例子整合了第一个和第二个故事里面用的两种方法。这是来自格拉斯哥大学的Andrew Tobin教授和Sosei-Heptares公司Malcolm Weir博士团队以及多个团队的合作。在这个工作中他们基于结构开发了一个M1乙酰胆碱受体的特异性激动剂,这个化合物具有治疗老年痴呆症(AD)的潜力。因为人的记忆和大脑内的胆碱能系统有关,提升脑内的乙酰胆碱水平可能改善老年痴呆症患者的认知能力。临床用于治疗AD患者的药物中有一类是乙酰胆碱酶抑制剂,如多奈哌齐(donepezil)。这类药物通过抑制乙酰胆碱的水解,提升体内整体乙酰胆碱的水平,在早期AD患者中有一定疗效。但是乙酰胆碱酶抑制剂药物在使用中存在明显的剂量依赖的副作用,尤其是胃肠系统副作用。这些副作用限制了它们的临床应用。这是因为这类药物通过提升体内整体乙酰胆碱水平,同时激活人体所有的乙酰胆碱受体,而有些受体的激活会引起副作用。人体内一共有5类乙酰胆碱受体:M1,M2,M3,M4和M5。其中M1受体是治疗老年痴呆的主要靶点,而M2和M3受体是主要的副作用靶点。因此研究人员希望开发特异性激活M1受体的激动剂。在这个工作中,研究人员基于M1受体的结构模型,通过分子对接虚拟筛选了具有新骨架的M1受体激动剂,并基于结构对其进行了优化,最终得到一个具有良好药理性质的M1激动剂HLT9936。这个化合物可以顺利穿过血脑屏障,在老鼠和比格犬等动物模型上表现出改善认知的疗效,并且在健康老年志愿者身上展示出激活学习和记忆中心的效果[17]。进一步的临床实验正在进行中。
来自学术论文的基于结构指导药物设计的例子还有很多,很多公司也在尝试利用结构指导GPCR药物的优化。Christ Tate教授2020年在Cell杂志发表综述总结GPCR结构对药物设计的影响[18]。如他在文章中所说:通常一个药物从研发立项到获批上市需要10-15年的时间。Rhodopsin之外第一个高分辨率的GPCR结构的出现只是14年前的事情(2007年)。因此现在评估GPCR结构对于指导药物研发直至药物成功上市的影响还为时尚早。但从阶段性进展来看,GPCR的结构信息对于设计更好的药物具有重要的指导价值。值得一提的是虽然目前小分子药物约有三分之一靶向GPCR,这些所有靶向GPCR的临床药物只涵盖了约100多个GPCR靶点。人体内有800多个GPCR,除去其中一半是负责嗅觉,剩下的400个里面,还有300个左右GPCR属于潜在的药物开发靶点。针对这些GPCR的结构研究和药物筛选很有可能在将来某一天为人类带来更好的靶向药物。这也是为什么我们持续对研究GPCR的结构和功能感兴趣的原因。
作者贡献:
本文由刘翔宇执笔,任引航和张翔协助参与文章中部分内容的背景检索。图1由张欣手画。
作者刘翔宇
刘翔宇,分别于2004年和2011年获得北京大学学士学位和博士学位,后加入诺奖得主Brian K. Kobilka 教授实验室(清华)从事博士后研究,现为清华大学药学院助理教授、清华大学结构生物学高精尖创新中心研究员。2012-2013年荣获清华北大生命科学联合中心博士后基金,2013-2017年获得安进中国博士后奖学金,2021年获得国家自然科学基金委员会优秀青年基金支持。他的主要研究领域为与疾病相关的G 蛋白偶联受体(简称:GPCR)的结构生物学研究,以及基于结构的药物设计。GPCR作为人类基因组编码的最大类别膜蛋白超家族,有800多个家族成员,与人体生理代谢几乎各个方面密切相关。
参考文献
[1] Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B. & Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov 16, 829-842, doi:10.1038/nrd.2017.178 (2017).
[2] Henderson, R. et al. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol 213, 899-929, doi:10.1016/S0022-2836(05)80271-2 (1990).
[3] Grigorieff, N., Ceska, T. A., Downing, K. H., Baldwin, J. M. & Henderson, R. Electron-crystallographic refinement of the structure of bacteriorhodopsin. J Mol Biol 259, 393-421, doi:10.1006/jmbi.1996.0328 (1996).
[4] Kimura, Y. et al. Surface of bacteriorhodopsin revealed by high-resolution electron crystallography. Nature 389, 206-211, doi:10.1038/38323 (1997).
[5] Pebay-Peyroula, E., Rummel, G., Rosenbusch, J. P. & Landau, E. M. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science 277, 1676-1681, doi:10.1126/science.277.5332.1676 (1997).[6] Palczewski, K. et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739-745, doi:10.1126/science.289.5480.739 (2000).
[7] Lefkowitz, R. J. & Hall, R. A funny thing happened on the way to Stockholm. (Pegasus Books, Ltd., 2021).
[8] Kobilka, B. K. Amino and carboxyl terminal modifications to facilitate the production and purification of a G protein-coupled receptor. Anal Biochem 231, 269-271, doi:10.1006/abio.1995.1533 (1995).
[9] Serrano-Vega, M. J., Magnani, F., Shibata, Y. & Tate, C. G. Conformational thermostabilization of the beta1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci U S A 105, 877-882, doi:10.1073/pnas.0711253105 (2008).
[10] Warne, T. et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454, 486-491, doi:10.1038/nature07101 (2008).
[11] Rasmussen, S. G. et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 450, 383-387, doi:10.1038/nature06325 (2007).
[12] Rosenbaum, D. M. et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266-1273, doi:10.1126/science.1150609 (2007).
[13] Cherezov, V. et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258-1265, doi:10.1126/science.1150577 (2007).
[14] Rasmussen, S. G. et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549-555, doi:10.1038/nature10361 (2011).
[15] Kang, Y. et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561-567, doi:10.1038/nature14656 (2015).
[16] Liu, H. et al. Structure-guided development of selective M3 muscarinic acetylcholine receptor antagonists. Proc Natl Acad Sci U S A 115, 12046-12050, doi:10.1073/pnas.1813988115 (2018).
[17] Brown, A. J. H. et al. From structure to clinic: Design of a muscarinic M1 receptor agonist with potential to treatment of Alzheimer's disease. Cell 184, 5886-5901 e5822, doi:10.1016/j.cell.2021.11.001 (2021).
[18] Congreve, M., de Graaf, C., Swain, N. A. & Tate, C. G. Impact of GPCR Structures on Drug Discovery. Cell 181, 81-91, doi:10.1016/j.cell.2020.03.003 (2020).
本文经授权转载自微信公众号“结构生物学高精尖创新中心”。

0
推荐
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}