对国产药的评价需要回归客观公正的环境。
撰文丨周叶斌(美国阿拉巴马大学伯明翰分校博士)
自11月11日国务院公布了优化疫情防控的“二十条”之后,许多人开始个人储备药物,以应对防疫政策宽松后可能面临的感染风险。11月18日,网上有药店公开销售国产抗新冠口服药阿兹夫定片。然而,不到24小时,阿兹夫定片已紧急全网下架,厂家称阿兹夫定是处方药,不适合在家自行服用。这一“上网下网”风波,引起了大家的关注。
阿兹夫定(Azvudine,FNC)是河南真实生物科技有限公司(简称“真实生物”)研制、国内首款获批的治疗新冠病毒肺炎的小分子口服创新药。2021年7月20日,阿兹夫定以抗艾滋病药物通过中国国家药监局附条件批准,老药新用,又于今年7月25日获药监局附条件批准上市,用于治疗新型冠状病毒适应症,并被纳入《新型冠状病毒肺炎诊疗方案(第九版)》。阿兹夫定获批治疗新冠近四个月后忽然可以公开网购,又忽然下架,引发了网上“买到了能不能吃”的焦虑,这提醒着我们去关注、了解这款国产新药的相关信息。
情况不容乐观。
1、仍未发表的数据
阿兹夫定作为抗新冠口服药获批是通过紧急使用授权,从未公布过完整的临床试验数据。当然,这并不能阻止药企宣称数据已上报,正在整理,准备投到国际期刊发表。不过这些话是今年7月下旬获批时说的,如今已然岁末,阿兹夫定要在国际期刊公布的临床试验数据在哪里?
其实投稿国际期刊与公示数据没有任何矛盾,研究人员完全可以将试验结果以预印版形式先公开。自新冠疫情暴发以来,我们看到无数重要的新冠研究先以预印版论文公布,再发表于期刊,生命科学各大顶级期刊无不接受这一操作流程。然而,三个多月来,除了打算建一年几十亿剂的生产线外,阿兹夫定未曾公开过任何数据。
我们能找到的也仅有8月相关药企申请港股IPO的材料中极为有限的数据[1]。

就是这些IPO文件里极为简略的内容,仍然让阿兹夫定看上去问题多多。本文分析如下。
2、试验人数问题
在IPO文件里提到阿兹夫定有三个临床试验,分别在中国、俄罗斯与巴西进行,中国的已经完成,俄罗斯的也达到了设计的招募人数,而巴西的只完成了招募人数的一半[1]。
中国的试验从2020年6月做到了2022年3月,计划招募342人,实际招募348人,标准是轻症与普通型新冠。俄罗斯的试验从2021年6月开始,计划招募314人,已招募314人,标准是中症。巴西的试验与俄罗斯一样,在2021年6月开始,标准是中症,计划招募342人,但只完成招募180人。
这些试验的主要终点不一,中国的是载毒量下降,俄罗斯与巴西是症状缓解。但无论什么终点,计划招募人数如此之少都是非比寻常的。
有人可能会说,管它招募多少人,最后有效性指标——临床试验终点——能做出统计意义上的区别不就行了?可是要在非常少的招募人数下做出显著差异,意味着药效要非常好,这样用药组与安慰剂组才能拉开足够的差距。但三期临床试验是前瞻性研究,试验完成前(包括设计试验的时候),没人知道药效有多高。因此试验人数的确定,一般是在“希望能有多大概率(统计检验功效)确认至少多高的有效性”这一基础上去推算的。
比如新冠疫苗的临床试验,假设希望能有90%的把握确认一个50%有效性的疫苗,可以回推需要多少病例,再根据一些感染率假设,推算应招募多少人以及试验需要做多久。
新冠药物在试验设计阶段,认为只要招募三百多人就能确定有效性,几乎是匪夷所思的。
我们可以参考获得FDA批准的辉瑞与默克两家的口服药。在轻到中症的高危人群三期临床,辉瑞计划招募约3000人,希望有1700人有数据做主要分析[2],默克则计划招募1550人[3]。这些都是基于“希望有足够的统计检验功效去检测50%降低重症风险”而演算出来的需要的样本量大小。最后两家实际分析的数据量分别有2200多人与1400多人。
即使说这两个药检测的降低重症风险与阿兹夫定的临床试验终点不同,那参考辉瑞口服药在低危人群的EPIC-SR试验——这里主要终点是症状持续改善——也招募了1440人[4]。
国内新冠单抗药Brii-196/198,参与NIH的ACTIV-2试验,确认有效性是用药组418人,安慰剂组419人[5]。
为什么阿兹夫定的研发方认为300多人的试验就能验证药物有效性?同行都是计划招募一两千人来明确有效性的时候,有人却不断设计300多人的临床试验,这是需要警惕的。
3、魔鬼在细节
阿兹夫定药企的IPO文书中,中国与俄罗斯两项临床试验的阳性结果有很多细节值得关注,以下仅举几例。
中国的三期临床试验中,主要终点是受试者服药后第7与14天时的载毒量。可是在描述有效性时,文件中却加了一个前提——基线载毒量高于310,在这些高载毒量受试者中,第3、5、7天用药组载毒量下降比安慰剂组更多[1]:

那么问题来了,这载毒量高的受试者是多少人呢?这一标准是事先确定的,还是事后加入?如果是事后加入,是否存在偏倚(bias)呢?而且即使是在这不知道多少人的高载毒量组里,载毒量变化达到显著差异的只有第5天。
根据这些描述可以推断试验的主要终点——受试者第7与14天的载毒量,用药组与安慰剂组没有显著差异,也就是说该试验没有达到主要终点。
另外,IPO文件里还提到所有次级终点均未显示显著差异。那么次级终点里有什么呢?除了吸氧比例、肺炎变化等症状,还有核酸检测转阴时间与速率。这与阿兹夫定获批时的新闻稿里“5天核酸转阴”可是直接矛盾的。姑且不说新闻稿是否准确,核酸检测结果与载毒量是有一定对应关系的,如果核酸转阴时间、速率在用药组都无改善,所谓基线高载毒量组第5天载毒量下降更多,这一阳性结果能有多靠谱呢?
再来看俄罗斯的试验,这个试验也是“阿兹夫定有助症状缓解”的说法来源[1]:

根据药企的说法,第7天时,用药组40.43%的人症状有改善,而安慰剂组只有10.87%,差异非常大。可这是PPS分析,是去掉一些随机入组但不符合试验流程的受试者之后所做的分析。实际这个试验两组都已经招募了157人,但PPS分析时分别只有141与138人,其它的人是因为什么原因被去除?如果没被去除,结果是如何呢?
一般来说,PPS展示的是理想情况下药物的有效性,因为各种不符合试验理想流程的人都可以剔除。而ITT(意愿治疗分析)与FAS(全分析集)更贴近最初随机入组的人群,也更严格。要知道三期临床试验能作为检验新药有效性的金标准,很大一个原因是受试者分配入组是随机的,而这个随机是在招募时确定的。ITT就是真实反映了这个随机状态,而PPS涉及剔除掉任何“不符合试验流程的受试者”,在保存试验的随机性上不如ITT或FAS(ITT中去掉完全没用药等极特殊情况)。
但是,无论是中国试验还是俄罗斯试验,IPO文件都未提供ITT或FAS的分析结果。中国试验里提到了FAS人数,可给出具体数据的却只是高载毒量组这样一个亚组分析,对“随机”背离更远了。
当一个药企只提供亚组分析、PPS分析这类更宽松、潜在偏倚风险更大的结果,而没有ITT、FAS数据,甚至连亚组与PPS具体标准都没有时,我们必须要高度警觉。
此外,在俄罗斯试验里,第7天用药组(下图蓝线)符合症状缓解标准的已经达到40%,可中位症状缓解时间却是10天,也就是说第10天用药组符合缓解标准的是50%。前7天与8-10这三天的缓解速率差异不小。安慰剂组(下图橙色)则反了过来,第7天——也就是第一周还只有10%的人缓解,再过一周(第13天)却有一半人缓解。走势如下:
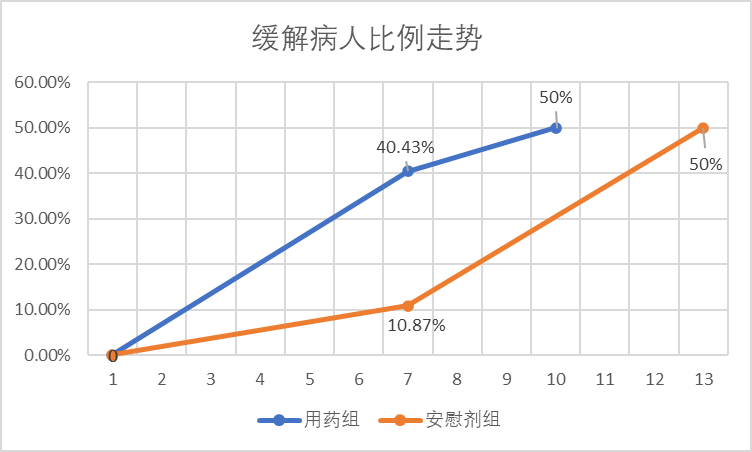
作者根据IPO数据绘制
缓解病人占比的速率如此急转也是较为罕见的情况,需要更详细的数据公开,才能搞清楚其中的原因。但IPO提供的数据不够详实,无法对此做出任何解释。
4、老药就安全吗?
IPO材料的信息透明度低,令阿兹夫定的有效性显得极为可疑。不过,在关注有效性问题之外,我们更需要关注药物的安全性问题。
文章开头提到,阿兹夫定在2021年7月获批用于HIV治疗。有人说,阿兹夫定是老药了,它的安全性是有保证的。但要知道,阿兹夫定作为抗HIV药物,获批依据是一个事先未设定标准的二期临床试验的事后非劣性分析[6]。
在已经有大量高效HIV抗病毒药上市的情况下,为什么阿兹夫定能获得如此“优待”,可以附条件优先审批?在没有大规模三期临床试验数据的情况下,只有295人使用过至少一剂治疗就被允许上市[6]?
阿兹夫定最初作为抗HIV药物时,对标的是拉米夫定(Lamivudine,3TC)。去搜阿兹夫定国内的报道,到处是“比拉米夫定剂量低”“不受拉米夫定耐药性影响”的溢美之词。
但是这些一边倒的正面报道并没有告诉我们,拉米夫定的遗传毒性实验结果[10]除了一个细胞实验结果不明确,在体内体外均为阴性,而阿兹夫定则在体内体外均为阳性(下文会讲到);拉米夫定的致癌性实验结果为阴性,而阿兹夫定根本就缺乏致癌性实验数据,还得补上。

拉米夫定的遗传毒性实验结果[10]
至于耐药性,阿兹夫定的HIV临床试验里也出现了耐药性突变[6]。不受拉米夫定的耐药性突变影响,不代表HIV病毒不会对阿兹夫定产生耐药性,只不过发生不同的耐药突变而已。
当我们给中国的HIV感染者使用阿兹夫定而不是拉米夫定的时候,他(她)们知不知道这些差异,还是只听说阿兹夫定比拉米夫定更好?他(她)们最基本的知情权有没有被尊重?
5、遗传毒性
前文提到,阿兹夫定获批抗HIV药物根据的是一个临床二期非劣性试验结果。也多亏一年前的这个批准,我们能找到当时药品监管机构的技术评审报告[6]。
该报告提到,阿兹夫定在遗传毒性实验里“Ames 试验、CHL 染色体畸变试验和体内小鼠微核试验结果均为阳性”:

遗传毒性(genotoxicity)是某个化学物质导致细胞内遗传物质发生突变的能力。对于一个药物,我们需要评估其遗传毒性,因为万一它能导致人体细胞的基因组发生突变,那就有潜在的致癌性、致畸性等严重安全问题。
遗传毒性不是一刀切的风险。这体现在两个层面。评估遗传毒性的方法有很多,比如用实验室培养的细胞,看看药品有没有在这些培养的细胞里导致突变,也可以在动物实验里观察有没有突变发生;而同一个药物用不同实验,做出来的结果又可能不一样。第一个层面就是说我们只能综合考虑,给出一个风险上的评价。
从不同检测方法的结果来看,阿兹夫定的评审报告包括了三种常用的遗传毒性评估方法:①Ames是用细菌分析致突变性,②CHL染色体畸形是用小鼠细胞观察,③体内小鼠微核试验则是在动物身上直接研究。三者与人体的相关性也有一定的递进关系,细菌是原核生物,与人体细胞相差甚远,小鼠细胞就接近很多,但体外培养细胞与真实的药物使用还是有很大差异,到小鼠体内试验,基本是遗传毒性在实验阶段能与人最接近的情况了。
这三项评估全部是阳性,也就是三种试验都能观察到阿兹夫定的致基因组突变现象,遗传毒性可说是不容忽视。
遗传毒性还要在另一个层面考虑,那就是药品的风险收益。如果是不治之症,那么对遗传毒性的风险耐受度会更高,也就是对药物的毒性标准会更宽容。毕竟,假如没有其它替代药物,那也不得不宽容。
到阿兹夫定的新冠治疗上,遗传毒性无论在哪个层面好像都不该宽容。参考同样遇到潜在致突变风险的默克口服药莫那匹韦(molnupiravir):在FDA上市审核时,遗传毒性成了这个药重点讨论的话题(详见《全球首款抗新冠病毒口服药莫那匹韦,是天使,还是魔鬼?》)。实际上,在多种实验里,莫那匹韦只有一个Ames实验阳性[7],动物体内实验都是阴性(见下图)。即便如此,FDA仍然对莫那匹韦做出了极为严格的限制——孕妇与未成年人直接排除在外,剩下的也必须在其它药物——包括paxlovid和单克隆抗体——都不能用的情况下使用。

莫那匹韦是全球首款抗新冠口服药。尽管如此,在其接受审核时,FDA多位专家提出,一旦有其它更好的药物上市,就需要考虑撤销莫那匹韦。此后,paxlovid横空出世(详见《无惧变异:辉瑞新药Paxlovid或将破除新冠阴影》),莫那匹韦的使用范围的确变得非常狭窄了。
现在阿兹夫定的遗传毒性风险比莫那匹韦明确得多,药物疗效证据却更少。莫那匹韦当时是一枝独秀,且的确可以有效降低重症死亡风险,但现在市面上已经有多款其它抗病毒药与单克隆抗体,阿兹夫定的上市还有必要吗?
6、生殖毒性
阿兹夫定还存在明显的生殖毒性风险。大鼠与兔两种常用生殖毒性实验动物中,有多个结果阳性[6]。
比如测试大鼠生育毒性:在交配前后给大鼠用药,无论是给雌性还是雄性大鼠喂药,高剂量时都会导致生育力下降:

阿兹夫定对胚胎发育也有潜在风险。在大鼠妊娠早期给药,会导致胚胎丢失率上升,5mg/kg的高剂量还会影响胚胎骨骼发育。
大鼠妊娠后期给药则对母体与子代均有毒性,子代影响包括了一些生殖系统与神经系统影响:

这些结果在兔子的胚胎发育毒性实验里也可以见到,包括上述生殖系统影响、胚胎存活降低以及胚胎骨骼发育等问题。
一些人可能会说这些生殖毒性实验中所用剂量都比较高,比如大鼠妊娠晚期对母体有毒性的最低剂量(NOAEL)是0.5mg/kg,致死剂量是5mg/kg,而阿兹夫定治疗新冠的人体剂量才5mg,按体重算比这些实验剂量小很多。
但是,动物与人体在代谢水平上有差异,比如实验用的大鼠、兔子,体型比人小很多,代谢快很多。考虑药物剂量时需要做人体等效剂量的换算,比如,大鼠的剂量乘上0.162才是人体对应的剂量。这样算下来,大鼠妊娠晚期的最低有毒性剂量0.5mg/kg,对应人体是0.081mg/kg,按标准体重60千克算,人体剂量是4.86毫克,比治疗新冠用的5毫克还低。喜提零安全空间。
就算是雄性大鼠生育毒性的最低剂量高一点,5mg/kg,换算下来对应60千克的人,也就48.1毫克,跟治疗新冠的剂量比,不到10倍的安全空间。如果用药男性身形瘦小,只能说多保重了。
更重要的是,遗传毒性、生殖毒性自有其特殊性,不是所谓的“不良反应”,临床试验很难明确(更何况阿兹夫定HIV的临床试验是个二期非劣性试验,到目前为止所有HIV临床试验加在一起,用过阿兹夫定的人应该在300个以下)。临床试验里没观察到,就说没有,是很不严谨的。在药物研发过程中,正确的做法是参考动物毒性试验,一旦有,就认为是有潜在风险,就要考虑这类风险是否可接受。并不是说有毒性的剂量比较高,就要用较高的暴露剂量来探索潜在风险。
我们可以参考莫那匹韦的做法[2]:
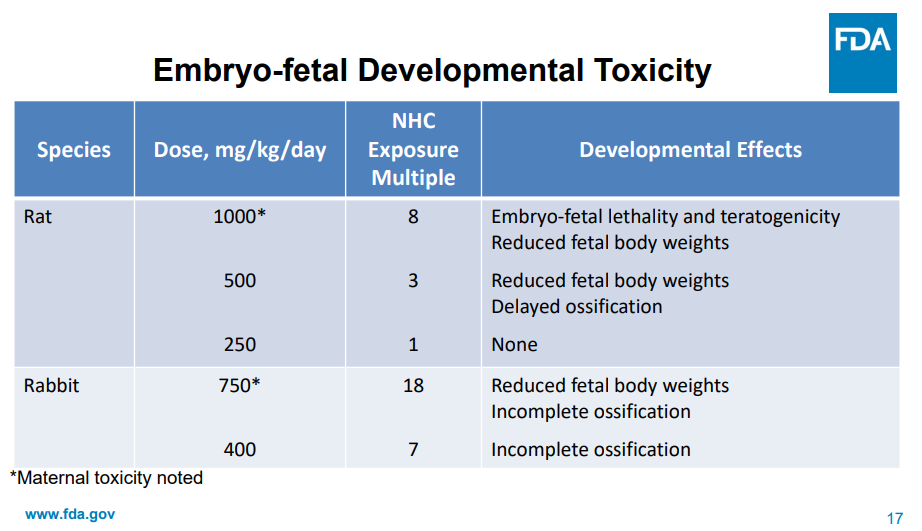
莫那匹韦的使用剂量是800mg,一天两次,但生殖毒性实验仍采用远超这一水平的剂量检测(在动物实验中需要更高剂量去观察,帮助明确这类风险)。莫那匹韦一样观察到了类似的胚胎发育毒性,特别是骨骼发育影响(倒没有阿兹夫定观察到的对母体毒性)。需要注意的是,因为有这些毒性,莫那匹韦被绝对排除在孕妇与未成年人中使用,还被摁死在了其它治疗药物均不可用情况下的最末位选择。
阿兹夫定的限制在哪里?它的遗传毒性类似于莫那匹韦,有效性却不如后者明确,这个药物在批准时是怎么考量的?
综上,阿兹夫定的安全性风险不是说我们一般说的常见不良反应或罕见不良反应,而是较为罕见的毒性问题。我们可以参考“吸烟致癌“——是否是每个抽烟的人都会得癌症?是否有一个明确的抽多少就会得癌症的标准?答案是没有。可是因为烟草致癌的风险是存在的,就需要把这个风险纳入管控的考量。药物的生殖毒性与遗传毒性类似,因为风险是存在的,就要纳入“这药能不能用”的权衡之中。
7、关键是总体风险
阿兹夫定针对HIV治疗的技术评审报告中还提到,致癌性实验尚未完成,需要补充[6]。考虑到这个药已经表现出非常明确的致突变性,也就是遗传毒性,致癌性的风险应该会是比较高的——癌变背后本身就是基因突变。
其实,更重要的是综合各项数据分析阿兹夫定的风险。
阿兹夫定(Azvudine,FNC)是核苷类逆转录酶抑制剂,化学结构上是核苷类似物,与胞嘧啶类似(核酸序列ATCG中的C)。另一个HIV常用药拉米夫定(Lamivudine,3TC)也是如此[8]:
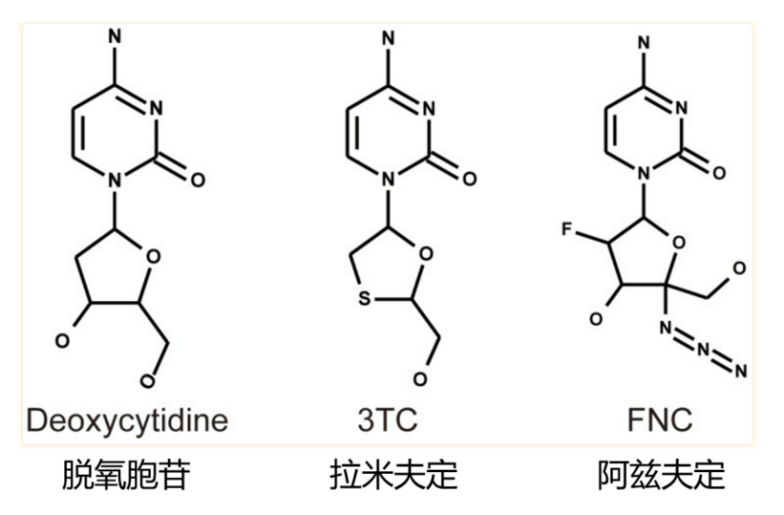
核苷类逆转录酶抑制剂是HIV药物里的一大类,它的结构与HIV逆转录复制基因组所需的原料——脱氧核糖核苷酸——类似,它与逆转录酶结合后可以阻断病毒复制。
但这样的机理必须关注遗传毒性与生殖毒性问题。因为万一结合的不是病毒的逆转录酶(对于新冠病毒来说就是RNA复制酶),而是人体DNA复制酶,影响了人体DNA的复制,那就有潜在的致突变风险。
不同的核苷类逆转录酶抑制剂的致突变风险不同。最早一个HIV药物齐多夫定(AZT)是致突变性最高的,很多后来者要安全得多。但每一个药物都需要独立分析。
对于阿兹夫定来说,目前的证据都表明它的遗传毒性、生殖毒性不可忽视。实际上还有其它研究指向阿兹夫定的这类风险。之前,有论文显示阿兹夫定可以抑制肿瘤细胞复制[9],抑制的机理是阻断了细胞分裂周期中DNA复制的那一阶段。既然肿瘤细胞一样是人的细胞,那么这意味着阿兹夫定针对的并不只是病毒的逆转录酶或RNA复制酶,而是同样会针对人体细胞的DNA复制酶。
更进一步来说,阿兹夫定抑制癌细胞复制的浓度并不高(微摩级别)——小鼠肿瘤模型的体内实验也在比较低的剂量(0.5mg/kg,对应人体剂量0.0405mg/kg,标准体重剂量2.43毫克,新冠治疗剂量的一半不到)就看到对人源肿瘤有抑制。这预示阿兹夫定影响人体DNA复制的效率并不低,更加证明了它有切实的遗传毒性。
作为一个新冠治疗药物,阿兹夫定的用药周期相对较短,这可能是对其遗传毒性、生殖毒性问题唯一的安慰。但问题是,我们需要综合考虑疾病的危险性、药物的有效性和安全性,以及其它备选药物是否存在。
目前,新冠病毒感染者绝大多数都可以自愈,特别是大量接种疫苗之后人群重症风险更低了。在这种情况下,口服药的安全性标准应该是非常高的。阿兹夫定表现出来的“症状缓解”是否还有实际意义?此外,现在国内已经有paxlovid与国产单克隆抗体药,这些药物有效性更明确,也没有严重的安全隐患。
08、药物安全不分国籍
从IPO文件来看,抗新冠药阿兹夫定的第一个人体临床试验最低剂量从1mg开始。根据动物毒性实验,最大无毒性剂量(NOAEL)对应人体是3mg,为了受试者的安全,一般人体试验起始剂量应该从NOAEL剂量的十分之一开始。如果有切实证据支持更高的起始剂量,那也不是不可以,可阿兹夫定有什么理由例外呢?
一些人很喜欢强调国产的重要性。但基本的药物有效性、安全性标准不应该因为药物的“国籍”而有松动。甚至对一些国产药应该更严才对,因为某些国产药说到底只会在中国上市,只有中国人会吃。对这些药的安全性有效性放松监管,谁来保护吃药的人?
对国产药的评价需要回归客观公正的环境,动辄扣国际领先、填补空白的帽子,是一种过度的“溺爱”,最后既害了国产药,也害了中国人。
参考文献
[1]
[2] [3] [4] [5] [6] [7] [8] [9] [10]
本文原标题为:有效性可疑,安全性堪忧,抗新冠口服药阿兹夫定是怎么上市的?

0
推荐
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}