阅读:0
听报道
文 | 药明康德
伊莎贝尔(Isabel)和安娜贝尔(Anabel)是一对混血双胞胎,她们融合了来自父母的德、日两国血统,却也不幸继承了父母的隐性囊性纤维化基因,这使得她们成为了世界上为数不多的囊性纤维化患者。自出生起这对姐妹就没有停止过与疾病的斗争,她们曾躺在生死线上静待下一刻的降临,也曾试图化解缺乏针对性药物和寿命缩短的绝望。
幸运的是,这一切都过去了。就在前不久,全球囊性纤维化患者迎来了历史性的一刻:具有治疗90%患者潜力的创新疗法Trikafta(elexacaftor/tezacaftor/ivacaftor和ivacaftor)获FDA批准上市。这一组合疗法也和“埃博拉药物初见成效”、“首张黑洞照片”、“神秘古人类”一同,被列为《科学》杂志2019年重磅科学新闻“年度突破”候选名单。

囊性纤维化(cystic fibrosis)是一种罕见的、缩短寿命的遗传性疾病,影响了北美、欧洲及澳洲约7.5万人的生命。疾病会在患者肺部等器官造成异常粘稠的粘液积聚,引起慢性肺部感染和进行性肺损伤,并最终导致死亡。在新药上市之前,由于大部分药物并不是对症下药,少部分针对病因的药物适用群体有限,囊性纤维化也曾被视为一种绝症。
好在绝症之下并非没有曙光。近半个多世纪来,从不明病因到对症下药,囊性纤维化患者预期寿命已经从10岁延长到了40岁以上。有人将人类与囊性纤维化的斗争视作对抗罕见病的医学奇迹,让我们一同回顾奇迹缔造之旅。
北欧传说中的“咸吻”之谜
在囊性纤维化作为一种疾病被承认之前,它所造成的影响常被归咎于不同原因,包括胰腺问题、呼吸道感染、营养不良等等。其中最令人匪夷所思的是北欧传说中关于婴儿的诅咒:当婴儿出生时,如果轻吻它的额头感觉很咸,那么这个婴儿是受到诅咒的,将很快死去。在当时,没有人能给出科学解释,更没有人料到这一现象将成为解密疾病的关键。
医学界对囊性纤维化的认识源于上世纪30年代末。美国病理学家多萝茜·安德森(Dorothy Andersen)在解剖营养不良儿童尸体的过程中观察到,一些患儿存在胰管扩张呈囊状和广泛纤维化现象,她将此描述为“胰腺囊性纤维化”,囊性纤维化由此被发现并得名。对此感兴趣的科学家们很快发现,囊性纤维化是一种常染色体隐性遗传疾病,并不只影响胰腺,还会引起包括消化道、肺脏在内人体多个器官病变,这也导致临床上常将囊性纤维化与其他疾病混淆。
1949年,一场突如其来的热浪给疾病研究带来了新的火种。正在医院照顾囊性纤维化患儿的儿科病理学家保罗·迪桑特·阿格涅斯(Paul di Sant’Agnese)观察到,一些病人出现了严重的中暑和脱水反应。进一步研究后,他找到了导致这一反应的根源——囊性纤维化患者汗液中的盐分显著高于正常人。这个意外发现让阿格涅斯医生不禁联想到了北欧传说中的“咸吻”,以此为灵感,他尝试用汗液测试替代插管定量测试胰酶来诊断囊性纤维化患者。这一方式减轻了年幼患者在疾病之外的痛苦和负担,很快在世界范围内普及并被不断改良沿用至今。

受阻的离子通道
阿格涅斯医生不仅为囊性纤维化患者带来了更便捷安全的诊断方式,也给疾病研究提供了思路,搞清楚患者汗液含盐量异常高的原因成为科学家们努力的新方向。保罗·昆顿(Paul Quinton)教授也是其中的一员,他甚至比大多数人更渴求真相,还不惜为此转移了研究方向,因为他本人就是一位饱受疾病折磨的囊性纤维化患者。
在担任加州大学实验室研究员期间,昆顿教授从正常人和囊性纤维化患者处收集了大量的汗腺组织,通过检测比对两者钠离子和氯离子穿过腺体的能力,他有了惊人的发现——囊性纤维化患者汗腺上皮细胞的氯离子通道存在异常。
氯离子无法穿透皮肤上皮组织,导致汗液中的盐分无法重吸收,而盐分丢失往往伴随着更多水分的丢失,进而使患者在高温下更易出现中暑和脱水反应。另外,离子通道异常也使得许多器官细胞中盐和水的流入和流出不均衡,造成异常粘稠的粘液积聚堵塞管腔,容易引起呼吸道感染和营养不良,并最终导致死亡。
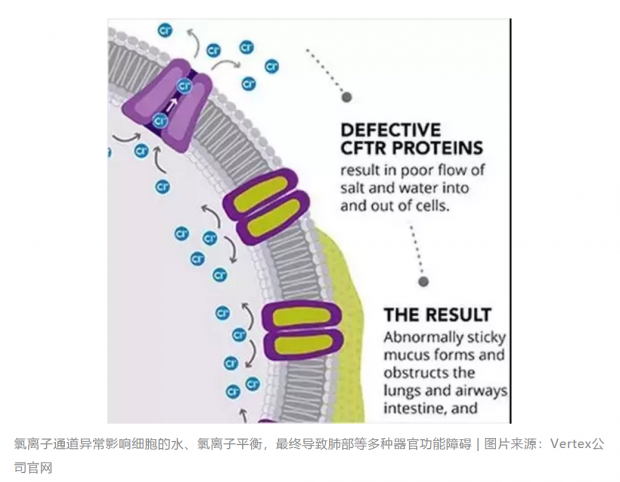
那是什么造成了囊性纤维化患者与生俱来的氯离子通道异常呢?上世纪80年代,一场基因层面的“寻宝竞赛”拉开了帷幕。此时,科学家对人类基因序列还知之甚少,致病基因的发现大多通过致病蛋白质或染色体异常,但这并不适用于囊性纤维化的研究。走投无路的科学家们想到了反向遗传学,即寻找基因组当中与疾病伴随遗传的标志物。到了1985年,利用反向遗传学,世界上有好几个科研小组发现囊性纤维化的致病基因位于人类第七号染色体上,但是这个染色体上有200多万个碱基,要从中发现致病基因无疑大海捞针。
徐立之教授和弗朗西斯·柯林斯(Francis Collins)教授同为“寻宝竞赛”大军中的一员,一次遗传学大会上的巧遇,促成了不在一个实验室的他们在囊性纤维化基因发现上的强强联手。在探索过程中,柯林斯教授的团队负责用染色体跳移的方法确定突变基因的大致位置,徐教授的团队负责在可疑区域内进行地毯式搜寻。两年的努力之下,他们的坚持获得了回报,《科学》杂志发表了他们的成果——异常的离子通道CFTR和造成囊性纤维化最常见的基因突变F508del终于被人类所发现。

一场没有保证的赌博

1989年8月25日,一个患有囊性纤维化的女孩在得知致病的缺陷基因被发现后,在日记上写下对未来的无限憧憬。但事与愿违的是,囊性纤维化的对症下药之路却走得漫长。当时,囊性纤维化的药物主要是帮助患者缓解症状、损伤和痛苦,并没有针对病因的药物诞生,而医药行业又很少涉足像囊性纤维化这样的罕见病药物研究。为此,时任囊性纤维化基金会总裁兼首席执行官罗伯特·贝尔(Robert Beall)博士提出了一个大胆的想法——与医药公司联手开发药物。
事实上,这并不是囊性纤维化基金会第一次在疾病研究中发挥力量。自1955年一群忧心忡忡的父母成立囊性纤维化基金会以来,这个以自己的力量拯救患病孩子的组织,就成为了科研之外的坚实后盾。无论是临床护理中心的完善,还是疾病登记注册制度的建立,基金会的出现都给无措的患者带来了极大帮助,甚至连囊性纤维化基因的发现也有基金会的支持。值得一提的是,命名囊性纤维化的安德森医生和发现患者汗液异常的迪桑特医生也是囊性纤维化基金会的早期发起人。

贝尔博士联系上了当时专门从事高通量筛选的Aurora生物科技公司(2001年被Vertex收购),提供7500万美元的经费用于针对囊性纤维化病因的小分子药物研发。随后,Aurora公司用当时先进的细胞检测平台对23万种药物进行了筛查,最终他们发现了一个可以针对CFTR蛋白上G551D突变位点发挥作用的药物。如果该位点发生突变,CFTR蛋白结构的打开速度就会变慢,而这种名为VX-770的复合物恰好可以帮助打开跨膜通道。
一项研究表明VX-770可以减少患者呼吸道感染的发作次数,并改善呼吸功能。2012年,仅仅经历短短3个月的评审工作,FDA就批准了首个针对囊性纤维化病因研发的药物Kalydeco(ivacaftor,即VX-770)上市,用于治疗携带G551D基因突变的囊性纤维化患者。虽然只有大约4%的患者能从中获益,但新药的上市还是给人们带来了信心,它不仅证明了靶向CFTR蛋白治疗囊性纤维化是一条可行之路,更有意义的是,它变革了罕见病药物的开发方式。
书写下一个篇章
此后,在基金会和医药公司的通力合作下,惠及更广泛囊性纤维化患者群体的药物稳步登场。2015年7月,FDA批准了囊性纤维化的第一种联合治疗Orkambi(ivacaftor/lumacaftor)上市,用于治疗6岁及以上拥有两个F508del突变拷贝的囊性纤维化患者。就在前不久,FDA只用了不到3个月的时间就批准创新疗法Trikafta(elexacaftor/tezacaftor/ivacaftor和ivacaftor)上市,用于治疗12岁以上至少携带一个F508del基因突变的囊性纤维化患者。

Trikafta由三种有效成分构成,其中elexacaftor是新一代CFTR蛋白矫正剂,它用于恢复携带F508del突变的CFTR蛋白的功能,从而改善患者的呼吸功能。Tezacaftor可以通过增加CFTR蛋白转运到细胞表面的水平来增强CFTR蛋白功能,而ivacaftor可以通过延长细胞表面CFTR蛋白的开放时间来提高缺陷型CFTR蛋白的功能。Vertex公司主席、总裁兼首席执行官Jdffrey Leiden博士在采访中这么描述它,这是一款具有治疗90%囊性纤维化患者潜力的突破性疗法。
在“重塑”CFTR功能的突破性疗法之外,科学家也找到了囊性纤维化药物研发的新路径。两性霉素B是一种由细菌产生的天然分子,在许多研究中,科学家们发现这种分子会在细胞上“打洞”,形成非选择性的离子通道造成破坏。因此,它一度被认为具有人体毒性。然而对于囊性纤维化患者来说,他们缺乏的恰恰就是离子通道。这就好像一间屋子的窗坏了打不开,污浊的空气不能及时排出。之前科学家们的研究方法要么尝试修复这扇窗,要么尝试在墙上安一面新窗。这次找到的小分子,则是简单粗暴地在墙上戳几个小洞,方便空气流通。
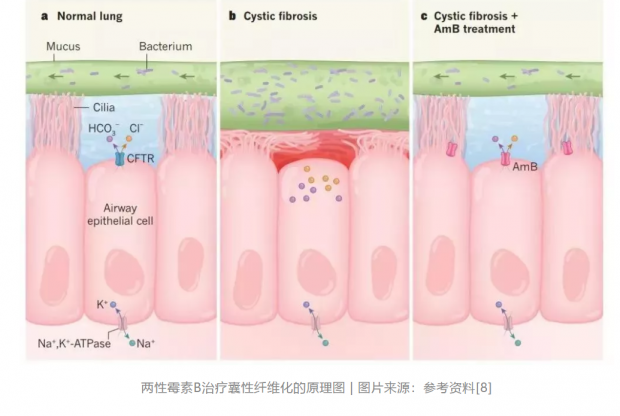
研究发现,在这款分子的作用下,细胞内的碳酸氢根离子能被顺利转运到细胞外,使呼吸道表面的黏液pH值恢复到正常水平,增强其杀菌能力。体外实验取得积极进展后,研究人员们又转而开展了体内实验。利用猪的疾病模型,研究人员们使用一款已经获批上市的两性霉素B,取得了同样的积极成果。这一发现也再次验证了这一潜在疗法的可行性。当然,在使用两性霉素B治疗囊性纤维化患者之前还有很长一段路要走。但它的出现还是让人们看到了一种全新疗法诞生的曙光。
2019年,是囊性纤维化突变基因发现的30周年,自1989年来的科学发现与药物转化把我们送到了与囊性纤维化对抗的关键时刻。与过去相比,囊性纤维化患者的生存状况得到了空前的改善,但这场抗争还没有结束,面对得不到药物帮助的囊性纤维化患者,能做的还有很多。就像现任囊性纤维化基金会主席普雷斯顿·坎贝尔(Preston Campbell)先生所期待的:“让我们永远心怀梦想,梦想我们的抗争之路终有尽头。直到有一天,我们可以说,我曾经是一名囊性纤维化患者。”
参考资料
[1] QUINTON, P. M. (1999). Physiological Basis of Cystic Fibrosis: A Historical Perspective. Physiological Reviews, 79(1), S3–S22. doi:10.1152/physrev.1999.79.1.s3
[2] Andersen, D. H. (1939). Cystic fibrosis of the pancreas, vitamin A deficiency, and bronchiectasis. The Journal of Pediatrics, 15(6), 763–771. doi:10.1016/s0022-
[3] ANDERSEN, D. H. (1946). CELIAC SYNDROME. American Journal of Diseases of Children, 72(1), 62. doi:10.1001/archpedi.1946.0202030006900
[4] Pearson, H. (2009). Human genetics: One gene, twenty years. Nature, 460(7252), 164–169. doi:10.1038/460164a
[5] Collins, F. S. (2019). Realizing the Dream of Molecularly Targeted Therapies for Cystic Fibrosis. New England Journal of Medicine. doi:10.1056/nejme1911602
[6] Kaiser, J. (2012). New Cystic Fibrosis Drug Offers Hope, at a Price. Science, 335(6069), 645–645. doi:10.1126/science.335.6069.645
[7] 速递 |重磅!仅用3月 FDA批准囊性纤维化突破性疗法,Retrieved Dec 4,2019 from
[8] 前沿 | Nature:大道至简!这种无药可治的疾病,意外找到了新药?Retrieved Dec 4,2019 from
[9] We Are at a Transformative Time in the History of CF, Retrieved Dec 4,2019 from
本文经授权转载自微信公众号“药明康德”。
话题:

0
推荐
财新博客版权声明:财新博客所发布文章及图片之版权属博主本人及/或相关权利人所有,未经博主及/或相关权利人单独授权,任何网站、平面媒体不得予以转载。财新网对相关媒体的网站信息内容转载授权并不包括财新博客的文章及图片。博客文章均为作者个人观点,不代表财新网的立场和观点。
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}