
撰文 | 史隽
4月20日 周一 MA累计确诊 39643例
上一篇日记讲到科学家们对新冠病毒进行全基因组测序,找到了西雅图和北加州新冠病毒传播的源头。
最近由纽约西奈山伊坎医学院 (Icahn School of Medicine at Mount Sinai) 和纽约大学医学院 (N.Y.U. Grossman School of Medicine) 的两个团队,分别通过测序分析3月中旬以来纽约的COVID-19患者身上的新冠病毒基因序列 (两个团队的样本不同),得出了基本相似的结论:新冠病毒在2月中旬就开始在纽约传播,病毒主要是从欧洲输入的,而不是亚洲。
然而,纽约州宣布的第一位确诊患者是一位从伊朗旅行回来的30多岁女性(3月1日确诊)。这意味着在第一例确诊患者被发现之前,新冠病毒已经默默地在纽约社区里传播开了。

图1:预印本文章
病毒侵入细胞后,会劫持细胞的分子机制来复制病毒。这个过程很快,但也很马虎。因此,病毒儿子可能会获得其祖先不存在的新突变。病毒儿子被释放感染别人以后,复制产生的病毒孙子也会继承这个突变,并且可能在此基础上增加新的突变。通过对病毒家族的全基因组进行测序,就能追踪病毒的突变过程。
等拿到很多的病毒家族成员的全基因组序列后,科学家们就可以用复杂的计算机程序分析比较突变,去观察随着子子孙孙从共同的祖先那里衍生出来,突变是如何一步一步地产生的。因为突变产生的速度大致是一定的,如果数据足够多,还能粗略的估计祖宗有多大年纪。
现在,世界各国的科学家们都在测序新收集到的病毒样本。因为疫情紧急,很多科学家会把尚未发表的病毒基因序列上传到名为GISAID的在线数据库,和全世界共享。
西雅图弗雷德·哈钦森癌症研究中心的Trevor Bedford(上期讲了他是怎么辟谣的:参见《117疫情观察:群体免疫还远着呢 | 史隽·Ⅵ》)等病毒进化专家团队则组织了一个名为Nextstrain的项目。当这些共享的数据出现在GSAID以后,科学家们就尽快去把这些序列下载到Nextstrain的后台,将这些序列纳入全球传播地图,并在上公布新冠病毒的基因组流行病学的最新信息,更新病毒家族树谱。
Nextstrain软件平台在过去几年逐步开发完善,曾应用于埃博拉、Zika和季节性流感等疫情,旨在让基因组流行病学在疫情期间尽快地发挥作用。
科学家们是怎么用病毒的序列来了解病毒传播方式的呢?
先介绍几个基本的概念。
有研究表明,新冠病毒的潜伏期短至2天,长至20多天。感染病毒5.1天以后,50%的人会出现症状;感染11.5天以后,97.5%的人会出现症状。
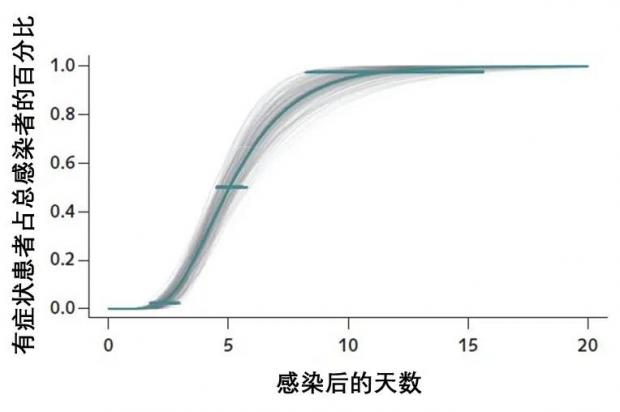
图2:来源
也有大型的研究证实,新冠病毒在潜伏期内就可以传染人,出现症状前1-3天有传染性。
因此,一个人感染后,在出现症状以前,就可能把病毒传染给另一个人。这个被传染的人也要过一定潜伏期,才会出现症状。这两个人出现症状之间的时间间隔被称为病毒的“世代间隔 (serial interval) ”。有研究估算新冠病毒的世代间隔期大约为7天。
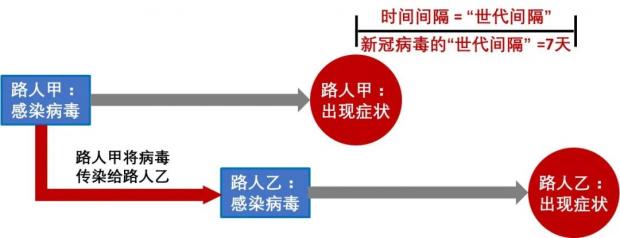
图3:病毒的世代间隔 (作者作图)
新冠病毒平均每月突变两次,也就是大约14天,2个世代间隔有一个突变。
简化的病毒传播链为如下所示:
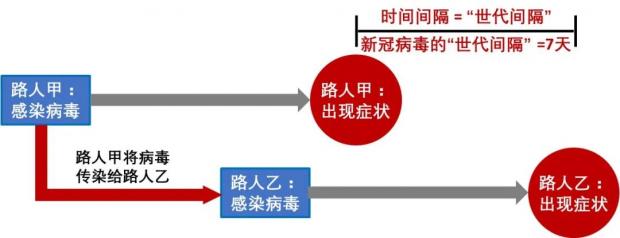
图4:病毒的传播链 (作者作图)
这些突变通常是很简单的一个碱基 “T”更改为“A”,或者可能是“G”更改为“C”。这些突变改变了病毒的遗传密码,不过这些小的变化一般不会令病毒产生关键变化,从而改变行为(如传染力和致病性)。
但是,科学家们可以结合取样时间和基因组的序列推断出感染的顺序。相比于传统的流调追踪接触者和病例分析,这种方法是一种更新的、了解病毒传播方式的办法。
举一个经典的例子,在《117疫情观察:新冠无症状感染的威胁 | 史隽·Ⅲ》里曾经提到过,Trevor Bedford通过病毒基因测序而发现无症状/轻症状感染导致西雅图地区社区传播。
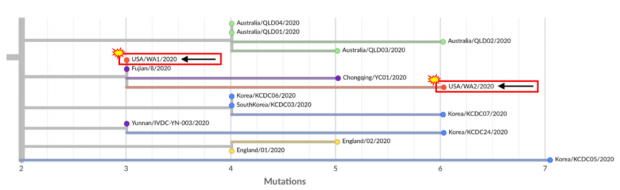
图5:来源于Trevor Bedford的博客 。
上图中的 “ USA / WA1 / 2020”就是美国第一例被确诊的COVID-19患者—— 1月15日从武汉返回西雅图Snohomish County (斯诺霍米什县) 的Amazon公司员工。他在1月19日被收集了鼻咽拭子样本,随后样本里的病毒被美国CDC快速测序了。
该病毒的基因组与1月21日在福建采样的“Fujian/ 8/2020“ 的基因组完全相同,福建的这个病毒也被标记是从武汉输入的,说明这两者很有可能是很接近的时间在武汉感染的。
“ USA / WA2 / 2020”就是上一篇日记(见《117疫情观察:群体免疫还远着呢 | 史隽·Ⅵ》)提到的西雅图流感计划 (Seattle Flu Study) 找到的西雅图地区最早的新冠病毒阳性样本。它是通过回顾测序西雅图地区从2020年1月1日至3月10日收集的有急性呼吸道感染的人的鼻拭子样本,而找到的2月21日采样的最早的新冠病毒阳性的样本。这个病例被认为是美国第二个社区传播感染的病例,取样于一名有轻微流感症状的少年。他在距离WA1病人的居住地约15英里处的一所高中上学。这位少年只有很轻微的症状,没有旅行史,并没有和WA1病人有任何接触。因此,他并不符合当时美国CDC测试新冠病毒的条件:“有发烧和呼吸系统症状,并且有中国武汉的旅行史”。患者因为有类似流感的症状就医,因为症状很轻,回家恢复了。
WA2病毒的基因序列和WA1的序列相比,几乎完全一样,只多了三个基因突变。
这个样本采集时间是2月21日,与第一个样本采集的1月19日间隔了差不多5周。根据上面图4的病毒传播链计算,5周的间隔时间也差不多可以累积2-3个突变。
WA1和WA2的病毒基因序列还包含一个关键的遗传变异 (在基因序列18060位置上),在数据库中59个来自武汉的样本里面,有2个含有这一突变。假设WA2不是WA1的后代,18060位置上的突变是随机产生的几率大约是2/59=3%。
换句话说,有97%的几率WA2病毒是WA1病毒的直接后代。考虑这两个病人活动的地区只间隔了24公里,这个可能性更大了。
这意味着,那位1月15日从武汉返回西雅图的男性在进医院就诊之前,已经把病毒传染给了别人,而那个人是无症状或者轻症状,没有引起警觉。从而导致新冠病毒在西雅图社区里面默默地传播了大约5周,感染了数百人。
后来的情况证实Trevor Bedford的结论是正确的:华盛顿州从后来的病人身上分离的病毒的基因组支持了第一二个病例(WA1和WA2)之间的关联。
再看另一个例子。
截至4月19日,Nextstrain里面有来自美国的新冠病毒的基因序列1149条,其中马萨诸塞州有19条,大部分都是美国CDC测序的。第一条“USA/MA1/2020”采样于1月29日,是那位从武汉回来的麻州大学的留学生,他的基因序列和来自武汉的病毒基因序列很相近。剩下的都是在3月4日至7日间采样,从时间上看很大可能是MA最早暴发的Biogen公司的员工样本 (参见《波士顿新冠暴发危机:Biogen年会病毒大传播始末》)。其中有一条孤零零的“USA/MA_1355/2020”样本采集于3月5日,和来自英国的病毒基因序列最接近。其他的17条形成一个大簇,和来自法国的病毒基因序列接近。这英法的区别不得不让我猜测:是因为两国社交礼仪的不同?
看起来,马萨诸塞州的早期病例大多是欧洲输入的。相比于马萨诸塞州超过36000的病例总量,这19个序列只是很少的一部分,相信以后还会有更多的序列被共享。
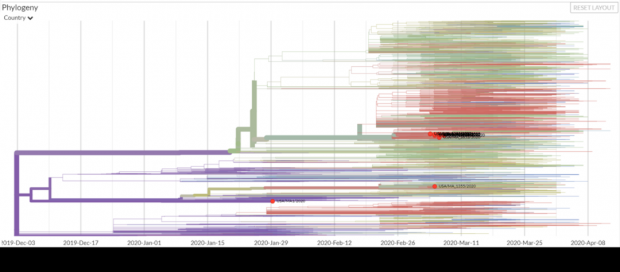
图6:Nextstarin截至4月19日全球新冠病毒树谱。图中标注的红点是马萨诸塞州的样本。来源:。
新冠病毒的变异速度比流感慢得多(后者大约是前者的2~4倍)。目前来看,病毒本身并没有很大的变异可以解释症状严重程度的不同。这对疫苗研发来说是一个好消息:“好不容易疫苗做出来,已经对付不了当前流行的病毒”,这种可能性降低了。但是,只要新冠病毒继续感染人,它就会继续突变。这就是为什么科学家需要时刻追踪病毒突变的原因——做好提前应对。

0
推荐
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}