阅读:0
听报道
你可能听说过一个创新药物从发现到最终上市可能需要花费十年以上的时间,耗费数十亿美元的投入,但始终不明白,为什么这么艰难?本文将为你详细介绍大名鼎鼎的抑郁症克星“百忧解”一波三折的研发历程。要突破哪些瓶颈?遭遇哪些失败?研究者经历多少倒霉事,咬碎几颗牙,才变出这一粒小小药丸?看完本篇,你会找到答案。
撰文 | 姬智
在精神病学发展的进程中,有一些发现至关重要,甚至从根本上改变了这个学科的面貌。比如弗洛伊德的精神分析学说;Ugo Cerletti发明的电抽搐治疗(Electroconvulsive therapy,简称“ECT”);John Cade发掘了锂盐作为精神科药物的应用价值等等。而在这些改变游戏规则的发现中,大名鼎鼎的“百忧解”(Prozac)无疑牢牢占据了一席之地。
百忧解,主要成分为氟西汀 (Fluoxetine),是第一个成功上市的选择性5-羟色胺 (5-HT) 再吸收抑制剂 (SSRI,Selective Serotonin Reuptake Inhibitors) 类抗抑郁药,其药物形态为盐酸氟西汀 (Fluoxetine hydrochloride) 。
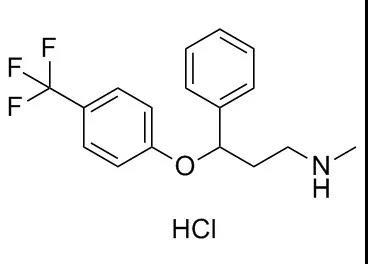
图1. 盐酸氟西汀化学结构
氟西汀由礼来(Eli Lilly)公司于1972年发现,1987年被美国FDA批准用于治疗抑郁症,給当时被抑郁症困扰的患者们带来了希望。也是从氟西汀开始,陆陆续续有SSRI类抗抑郁药上市,它们开辟了抑郁症药物治疗的新篇章,成为最广泛使用的抗抑郁类处方药。三十年悄然流逝,越来越多的基于新机制新靶点的抗抑郁药物登上市场舞台,但氟西汀在临床中依然十分常用,它是世界卫生组织所列出的基本药物之一,也是基本卫生系统必不可少的重要药品[1]。
抗结核药物的“副作用”
抗抑郁药物的研究起源最早要追溯到20世纪50年代先驱Nathan S. Kline的工作。Kline是纽约罗克兰县罗克兰医院的研究主任(Rockland Hospital in Rockland County,New York),当时结核病肆虐,临床医生们在使用抗结核药物治疗病人的过程中,意外发现了此类药物对病人有一种神奇的心理影响——它可以使病人表现出“不同寻常”的快乐。报道引起了Kline的注意,他开始进行自己的抑郁症药物研究,并与精神分析家Mortimer Ostow在1957年的美国精神病学协会年会上联合发表了这方面的报告[2],获得了当时学术界的认可。大家开始相信,药物治疗也能为抑郁症患者带来希望。自此,一种新的药物类别——抗抑郁药——诞生了。
早期的抗抑郁药物多为三环类抗抑郁药 (Tricyclic antidepressants,TCAs)。这类药物最早正是由抗结核药物改造而来,其核心结构为一个七元杂环,两边各连接一个苯环。它可以抑制突触前膜对去甲肾上腺素(NA)和五羟色胺(5-HT) 的再摄取,增加二者在突触间隙中的浓度,以延长它们作用于相应受体的时间,从而发挥抗抑郁作用。
三环类的代表药物有丙咪嗪、阿米替林、氯丙咪嗪、多塞平、地昔帕明、去甲替林、普罗替林等(图2)。其中阿米替林对伴失眠的抑郁患者疗效较好;丙咪嗪对内源性抑郁症、更年期抑郁症患者更为理想,对精神病的抑郁症状效果较差;而氯丙咪嗪是治疗强迫症的首选药物,同时也是美国 FDA 批准的治疗强迫症的药物之一;多塞平则对各类焦虑抑郁状态效果较好[3]。
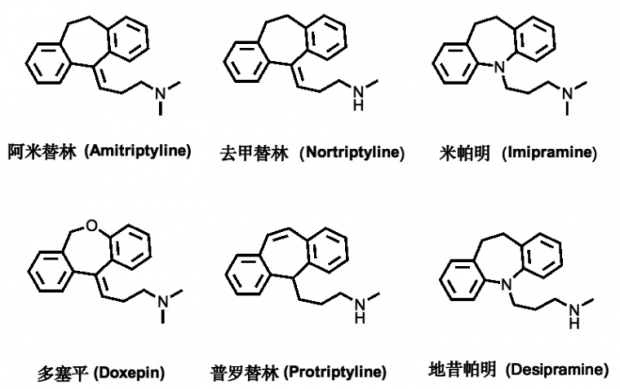
图2. 常见的三环类抗抑郁药 (Tricyclic antidepressants,TCAs)
三环类药物虽然被证实具有抗抑郁的效果,但也具有较为明显的毒副作用。例如,治疗安全窗小,有严重的心脏毒性,有抗胆碱作用(表现为口干、便秘、视物模糊、嗜睡、体重增加等),会引起体位性低血压等。因此,科学家们一直致力于研发毒副作用更小、疗效更好的药物。
生理学发展指明新方向
药物的发展离不开生理学研究的推动。早期分析抑郁症病人的尸检样本时,科学家们就已经发现,在自杀死亡的抑郁症病人后脑中,5-羟色胺(5-HT)和其代谢物 5-羟基-吲哚乙酸(5-HIAA)的水平要比突发性死亡或心肌梗死病人低得多[4, 5],这是否意味着人体内5-HT水平可能与抑郁症有关?
1969年,一本名为《神经组织的结构和功能》 (The Structure and Function of Nervous Tissue, Volume III: Biochemistry and Disease)的书出版,书中详细介绍了当时中枢神经生理学的“最新”知识[6],详细阐述了5-HT在人体内的合成、储存、释放、分解和再摄取的过程,这些过程符合5-HT作为神经递质的标准。同时,药理学也有证据证明,与NA相似,5-HT与抑郁症的病理密切相关。受此启发,科学家们转而开始寻找毒性较小的单胺递质再摄取抑制剂,期望通过特定地抑制5-HT的再摄取来改善抑郁症状。
现在我们知道,5-羟色胺(5-HT)也称作血清素(Serotonin),是一种单胺类神经递质。同属于这个家族的还有多巴胺(Dopamine, 简称“DA”)、去甲肾上腺素 (Norepinephrine,简称“NE”或“NA”) 等等,这些神经递质都能在神经元中合成,并能介导神经信号传递,因此也与人类的情绪调节有关。
随着生物学科的发展,现代设计新药往往基于已经确定的作用靶标,借助于强大的计算机辅助功能,来设计合成有作用的先导化合物。而回到1960年代,在当时生理学水平有限的情况下,科学家们能从抑郁症病人尸检样本分析中,敏锐地观察到5-HT水平与抑郁症的关联,并开创性地提出“选择性再摄取抑制剂”的概念,在当时确实是了不起的成就。
何为再摄取抑制剂?
5-HT在突触前神经元细胞中合成,释放进入突触间隙,与突触后神经元上的受体家族结合,进而介导神经信号传递。在此过程中,突触前神经元上的再摄取泵会将一部分游离的5-HT再次摄取,导致5-HT浓度降低。
SSRIs正是通过抑制再摄取泵来发挥作用,它使得突触间隙的5-HT浓度升高。相似的作用原理,三环类抗抑郁药可以同时抑制突触前膜对NA和5-HT的再摄取,不具有选择性;而氟西汀则只选择性抑制5-HT的再吸收,并不影响多巴胺与去甲肾上腺素的生理作用,这也是它的优势之一。
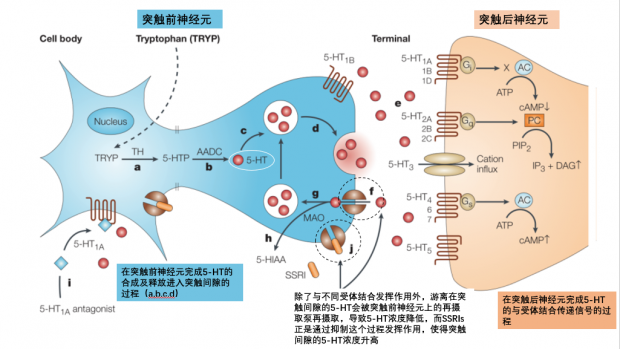
图3. 5-HT及SSRIs的作用机制【7】
不如人意的结构改造
1970年,礼来的药物化学家Bryan Molloy和药理学家Robert Rathburn开始合作,目的是开发一种新型抗抑郁药物,既能够保留三环类抗抑郁药物(TCAs)的治疗活性,又能够克服TCAs的心脏毒性和抗胆碱能作用。基于早期TCAs研究的基础,他们采取了药物诱导的小鼠体温降低模型对药物进行筛选,发现苯海拉明 (Diphenhydramine) 和其它一些抗组胺类药物也达到了和TCAs相同的效果。瑞典哥德堡大学(University of Goteborg)的Arvid Carlsson和同事们则观察到,苯海拉明除了拮抗组胺受体外,还能抑制单胺摄取,后者意味着它在治疗抑郁症方面可能会有效果[8]。
基于上述发现, Molloy对苯海拉明进行结构改造及优化,合成了许多类似物。在第一轮改造时,他们保持了含有N-甲基乙基胺的侧链不变,将苯海拉明的N,N-二甲基-2-(二苯基甲氧基)-乙胺结构演变为苯氧基苯胺母核,设计合成了一系列3-苯氧苯丙氨基化合物,并在Rathbun实验室对这批新合成的分子进行药理测试,发现化合物LY94939 (又称Nisoxetine,汉译“尼索西汀”) 具有与TCAs类抗抑郁药物相同活性。而之后的体外靶点活性测试表明,尼索西汀与地昔帕明一样,对去甲肾上腺素有再摄取抑制作用,而对5-羟色胺(5-HT)和多巴胺 (DA)的再摄取抑制作用则微弱很多[9, 10]。因此,科学家们继续探索,试图找到可以针对5-HT进行抑制的药物分子。
然而,这样的探索并不被看好,甚至受到了多方阻挠。例如,一位中枢神经系统研究方面的学术专家,在每季度访问礼来公司时,都劝他们打消追求单胺摄取选择性抑制剂的念头,因为当时其他科学家的观点是,增强5-HT的再摄取不仅无法治疗抑郁症,反而可能会导致抑郁。但研究团队没有放弃,幸亏有礼来CNS(Central Nervous System)研究委员会顾问Slater的支持,这个项目总算没有中断。
氟西汀:纤毫之差
我们必须知道的一个事实是,药物化学是一门非常精妙的学科,即使只有细微的结构差别,两个分子的药效也会有很大区别。在这个项目中,结构的细微变化也会改变分子对不同神经递质的选择。
1971年,另一位推动氟西汀诞生的重要人物——David T. Wong——加入了研究团队。Wong出生于中国香港,是当时著名的生物化学家,过去的科研经历让他十分熟悉5-HT对情绪调节的作用。在他的鼓励下,研究小组又设计合成了第二批50多个化合物,并将这两批化合物再次进行筛选,以寻找可能选择性抑制5-HT再摄取的分子。研究小组对这两批化合物重新进行了体外5-HT、NA、DA再摄取抑制活性测试,发现化合物LY82816(氟西汀的草酸盐形态)对5-HT再摄取表现出了非常好的选择性[11]。
其实,氟西汀与同一系列的化合物结构差别非常小。下图(图4)是这类化合物的构效关系总结,我们可以看出,3-苯氧苯丙氨基化合物中2个苯环均无取代时,其对5-HT和NA的再摄取抑制活性 (Ki) 分别为102nM和200nM。而在苯氧基对位引入三氟甲基 (即氟西汀) 后,对5-HT再摄取抑制活性提高了6倍 (Ki=17nM) 。不仅如此,正如上文所提到的,氟西汀还具有较强的靶点选择性,这意味着,氟西汀在抑制5-HT再摄取的同时,不会干扰其他神经递质的正常功能。
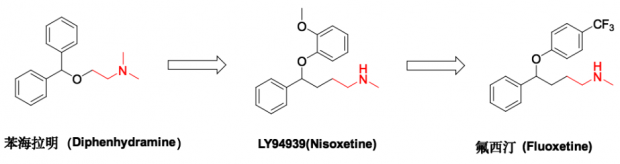
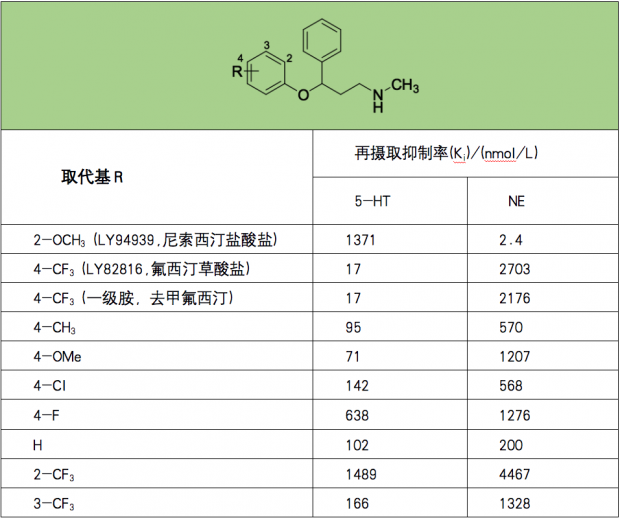
图4. 改造过程中系列化合物的构效关系
重建动物模型
谁知,与尼索西汀不同,氟西汀在药物诱导的小鼠体温降低模型中竟然没有显示出活性!原来,这一时期普遍采用的抗抑郁症研究的动物模型是建立在早期三环类抗抑郁药的基础上的,因而特别适用于抑制去甲肾上腺素(NA)再摄取类药物,但对氟西汀却无法显示出显著的体内活性。当务之急,研究者需要建立5-HT在体内的再摄取抑制模型。
经过不断摸索,Wong等科学家们建立了三种新的模型来评估氟西汀的抗抑郁活性。这三种动物模型分别是,小鼠强迫游泳实验 (Forced swim test);习得性无助模型 (learning helpless) 和嗅球切除模型 (Olfactory bulbectomy)。在这三种模型中,氟西汀都表现出了良好的效果,最终使大家认可了氟西汀。在此之后,这三种模型得到了广泛使用。
临床一波三折
最终,1976年,在动物体内的安全性实验全部完成之后,礼来公司向FDA提交了临床研究申请,首次对人服用氟西汀进行临床实验研究。一期临床进展非常顺利,氟西汀表现出良好的临床安全性,很快就准备好进入第二阶段:寻找治疗抑郁症患者的有效性指标。
在当时,礼来公司还有其他更靠前的候选药物需要考察,加之缺乏精神病学方面经验丰富的临床研究人员,氟西汀的临床二期试验一度陷入僵局。在资源有限的情况下,二期临床一开始只能以一个相对小的规模进行。然而,不幸的事再次发生了:在这一批小规模人群中,氟西汀被发现无效!
这给整个项目组带来了极大的打击。好在,研究小组经过分析之后,认为试验失败可能要归咎于志愿患者的类型——因为这批患者在先前的治疗中对其他抗抑郁疗法也没有反应。大家振作精神,继续推进,在另一群抑郁症患者中重新开始试验,终于取得了良好的结果。
1981年,氟西汀的临床III期试验开始,于1983年顺利完成。结果显示,氟西汀对抑郁症疗效显著,且副作用轻。这些试验的临床结果汇编在100多卷2英寸的活页夹中,提交给美国FDA,用于申请上市。从第一次人体给药,到完成所有的试验,提交材料,一共花费了七年多的时间[12]。
但是,对氟西汀和研究团队的考验还没有结束。
遭遇药物抢发
几乎同一时间,1982 年,阿斯利康制药公司(Astra Pharmaceuticals)在欧洲率先推出了治疗抑郁症的药物齐美利定 (Zimelidine)。无独有偶,齐美利定也是一种选择性 5-HT 再摄取抑制剂 (SSRI)[13]。礼来公司失去了第一个向市场推出SSRI的机会。
然而,齐美利定上市后不久,就曝出会引发一些罕见的副作用,尤其是引发类流感综合征。为了避免更大的损失,阿斯利康立即终止了有关 SSRI 的所有研究。这无疑送给了礼来一线生机。
1983年 10月,礼来公司研发团队代表与 FDA 咨询委员会会面,期望他们能综合评价氟西汀的临床研究结果及获批上市的可能性。两年后,也就是1985年10月,FDA咨询委员召开会议,讨论氟西汀的上市申请。虽然有人质疑氟西汀可能产生类似于齐美利定引发的流感样反应,但咨询委员会成员认为,两者结构有明显区别,这种特殊的副作用可能是齐美利定特有的,并一致建议批准使用氟西汀治疗抑郁症。
在研究团队焦急的等待中,1987年 12月29日,FDA 终于批准盐酸氟西汀上市。1988年1月,礼来公司以商品名“Prozac (百忧解)” 成功推出 5-HT 再摄取抑制剂氟西汀,用于治疗抑郁症(图4)。研究团队多年来承受的压力与质疑,终于在此刻被欢乐代替。
图5. 礼来公司成功推出 5-HT再摄取抑制剂氟西汀,商品名取为Prozac(百忧解)丨图片来源:
氟西汀疗效确切,副反应轻,上市之后广受欢迎,销售额连年增长,1988年全球销售额突破1亿美元,1992年突破10亿美元大关,成为第一个年销售额突破10亿美元大关的中枢神经系统药物。1999年销售额更是达到26.13亿美元,为全球第6畅销药品。《财富》杂志 (Fortune magazine) 将氟西汀列为“世纪药品”之一,2001年专利到期后,专利药市场销售额下降明显,但氟西汀的总处方量未受影响[14]。
氟西汀的一战成名给各大制药公司指引了新的方向,一大批公司加入5-HT再摄取抑制剂开发的行列,资金注入和研究人员的努力使得短短几年之内就有帕罗西汀、舍曲林、文拉法辛、西酞普兰等一系列SSRIs类抗抑郁药进入市场。给抑郁症患者带来了新的希望。
唯有信念
回到开头,为什么说百忧解的问世是改变精神病学发展进程的事件之一?除了它良好的抗抑郁效果外,更重要的是,百忧解在一定程度上重塑了公众对精神障碍的认识,重新定义了临床抑郁症的概念,将精神病学的方向从心理治疗扭转为生物学治疗。
由于百忧解非常安全,并且与其他药物之间相互作用的风险极小,所以之前不愿开具三环类药物处方的医生开始为症状较轻的患者使用药物治疗,这在一定程度上解除了当时抑郁症的污名化,令更多人敢于吐露自己的病情。因此,百忧解毫无争议地成为精神病学史上的标志性药物,其地位是不可撼动的。
氟西汀的成功问世也给科研人员们留下了宝贵的经验。研究者在16年的时间里历经各种考验,每一步都极为艰辛。他们的成功经验为后来的新药研发提供了非常好的借鉴意义:最初,没有建立合适的评价模型,导致氟西汀动物体内测试(人体外测试)无效;接着,公司优先发展其他药物,导致氟西汀临床研究暂停;同时,病人选择有误,导致临床研究失败;最后,同类药物副作用又导致公司决定暂停研发。如果不是研究人员坚韧的毅力和不懈追求真理的精神,氟西汀的研究可能早已夭折。而这种精神,在世人多急功近利的今天显得尤为可贵。

图6. 氟西汀研发过程中两位重要的科学家Bryan B. Molloy 与David T. Wong丨图片来源:左;右:)
科学的发展是一个不断积累的过程。神经科学的先驱们提供了生理学基础、开创性的设计思路,研究团队又通过深入的思考和不懈的努力来验证思路,遭受怀疑、嘲笑也不放弃,才最终取得了突破性的成果。就像David T. Wong在三十年以后回顾氟西汀的研发之路时说的那样:
It is essential that ideas are steadfastly championed by passionate believers to achieve the final goal.
参考资料
[1];jsessionid=9633BBC4D3838AC972A59899C442C306?sequence=1
[2] Loomer, H. P., Saunders, J. C., & Kline, N. S. (1957). A clinical and pharmacodynamic evaluation of iproniazid as a psychic energizer. Psychiatric Research Reports, 8, 129–141.
[3] Feighner J. P. (1999). Mechanism of action of antidepressant medications. The Journal of clinical psychiatry, 60 Suppl 4, 4–13.
[4] Shaw, D. M., Camps, F. E., & Eccleston, E. G. (1967). 5-Hydroxytryptamine in the hind-brain of depressive suicides. The British journal of psychiatry : the journal of mental science, 113(505), 1407–1411.
[5] Bourne HR, Bunney WE Jr, Colburn RW, et al. Noradrenaline, 5-hydroxytryptamine, and 5-hydroxyindoleacetic acid in hindbrains of suicidal patients. Lancet. 1968;2(7572):805-808.
[6] Bailey OT. The Structure and Function of Nervous Tissue, vol 3, Biochemistry and Disease. JAMA. 1970;212(9):1528–1529. doi:10.1001/jama.1970.03170220082030
[7] Wong DT, Perry KW, Bymaster FP. Case history: the discovery of fluoxetine hydrochloride (Prozac). Nat Rev Drug Discov. 2005;4(9):764-774. doi:10.1038/nrd1821
[8] Molloy, B. B., Wong, D. T . & Fuller, R. W. The discovery of fluoxetine. Pharmaceutical News 1, 6–10 (1994).
[9] Wong, D. T., Horng, J. S., & Bymaster, F. P. (1975). dl-N-methyl-3-(o-methoxyphenoxy)-3-phenylpropylamine hydrochloride, Lilly 94939, a potent inhibitor for uptake of norepinephrine into rat brain synaptosomes and heart. Life sciences, 17(5), 755–760. (75)90531-7
[10] Wong DT, Bymaster FP. Effect of nisoxetine on uptake of catecholamines in synaptosomes isolated from discrete regions of rat brain. Biochem Pharmacol. 1976;25(17):1979-1983. doi:10.1016/0006-2952(76)90053-8
[11] Wong DT, Bymaster FP, Horng JS, Molloy BB. A new selective inhibitor for uptake of serotonin into synaptosomes of rat brain: 3-(p-trifluoromethylphenoxy). N-methyl-3-phenylpropylamine. J Pharmacol Exp Ther 1975;193(3):804-11
[12] Wong DT, Bymaster FP, Reid LR, Threlkeld PG. Fluoxetine and two other serotonin uptake inhibitors without affinity for neuronal receptors. Biochem Pharmacol. 1983;32(7):1287-1293. doi:10.1016/0006-2952(83)90284-8
[13] Bengtsson BO, Wiholm BE, Myrhed M, Wålinder J. Adverse experiences during treatment with zimeldine on special licence in Sweden. Int Clin Psychopharmacol. 1994;9(1):55-61. doi:10.1097/00004850-199400910-00009
[14]
话题:

0
推荐
财新博客版权声明:财新博客所发布文章及图片之版权属博主本人及/或相关权利人所有,未经博主及/或相关权利人单独授权,任何网站、平面媒体不得予以转载。财新网对相关媒体的网站信息内容转载授权并不包括财新博客的文章及图片。博客文章均为作者个人观点,不代表财新网的立场和观点。
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}